
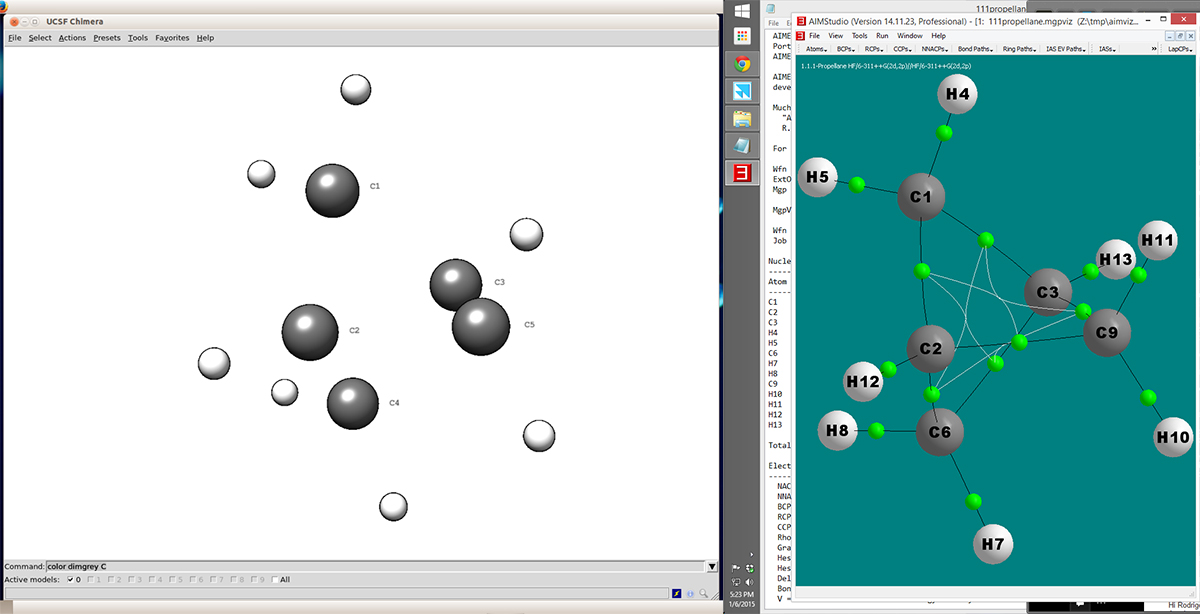
Hello! Thanks to the recommendations from Dr. Eric, I was able to code an extension for Chimera that reads mgpviz files generated from the AIMAll program. Right now, only the XYZ and atom type information is parsed and the molecule is displayed. Although, I do not know why it shows no connectivity. I am including a screenshot that shows how Chimera displays the molecule (left) and how does it look in AIMAll (right). I do use the command connectMolecule() but I do not know if I have to include some other argument for that function. I based the code from the ReadXYZ extension. My code (I am no coder, so it might be super ugly (: ) ChimeraExtension.py ------------------------------------------------------------------------------ def aimallOpen(fileName): from ReadAimall import readAimall return readAimall(fileName) import chimera chimera.fileInfo.register("AIMAll mgpviz", aimallOpen, [".mgpviz"], ["mpgviz"], category=chimera.FileInfo.STRUCTURE) the readAimall function: ----------------------------------------------------------------------------------- ## Extension to read a file output from AIMALL ## based on the code ReadXYZ def readAimall(fileName): from OpenSave import osOpen from chimera import UserError, Molecule, Element, openModels, replyobj from chimera import Coord, connectMolecule anums = {} ## Open the file getatoms = osOpen(fileName) ## Search for the string: ## 'Number of NACPs' ## which gives us the number of atoms ## and store it in numAtoms for data in getatoms: data = data.strip() if data.startswith("Number of NACPs"): a,b,c,d,e = data.split() numAtoms = int(e) getatoms.close() f = osOpen(fileName) readAtoms = 1 state = "init" ## Skip the 26 header lines from the mpgviz file f = f.readlines()[26:] ## cycle through the file until it reaches ## the number of Atoms for line in f: if readAtoms == numAtoms: break line = line.strip() if state == "init": state = "post" m = Molecule() r = m.newResidue("UNK", " ", 1, " ") state = "atoms" serial = 1 if not line: continue elem, charge, x, y, z = line.split() x, y, z = [float(c) for c in [x,y,z]] ## Remove the number from the atomic symbol elem = filter(lambda x: x.isalpha(), elem) element = Element(elem) anum = anums.get(element.name, 0) + 1 anums[element.name] = anum a = m.newAtom("%s%d" % (element.name, anum), element) r.addAtom(a) a.setCoord(Coord(x, y, z)) a.serialNumber = serial serial += 1 readAtoms += 1 #f.close() connectMolecule(m) return [m] --------------------------------------------------------------------------------------------------- I couple of questions: 1- Do I have to include something in the connectMolecule function to enable the display of bonds? 2- At the end, what would be your recommendation, based on your experience, to display the critical points and bonds between atoms? We were considering: Fake atoms, Markers, Sphere shapes and bond colors. What we are trying to achieve is to be able to turn off certain critical points and leave some on to show how molecules interact. Thank you for your help and let me know what you think!! Rodrigo.
{kind=link}