
On Mar 9, 2010, at 2:17 PM, Francesco Pietra wrote:
Hello Eric: I have seen your later correction mail.
I had a bad day, that is little time for chimera and with little success. Please try to guess were I was faulty.
francesco@tya64:~$ chimera --version chimera alpha version 1.5 (build 30193) 2010-03-06 09:27:34 GMT francesco@tya64:~$ This is amd64, at the moment I have no 32bit computer.
<select ligand> selects too much.
<select :residue.number> <select :residue.name> do select exactly what I want (which is a small ligand of HEME ligand of the protein.
The problem was with <define> command
MD-movie...Per-Frame "define centroid :residue.number" ... Apply
then activating the movie gave the same view as not going through the Per_Frame, i.e., the small ligand does not leave any sphere on the path. I did not investigate if the sphere are too small to be seen at the magnification I used, ore have the same color as the background (black). Very likely, I did some other major mistake.
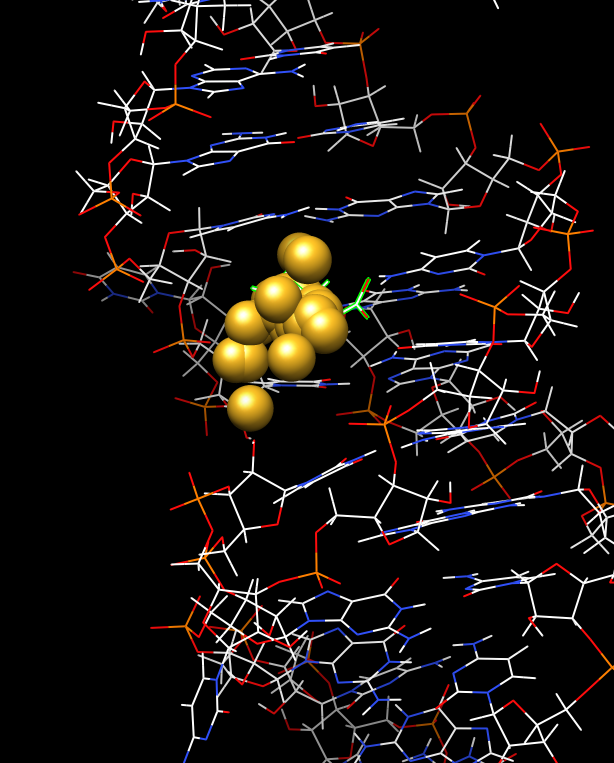
Not sure what to say. Works for me. I've appended an image of centroids tracing the motion of a small ligand in an RNA binding site. I used a step size of 10 and this 'define' command: define centroid raiseTool false radius 1.0 color goldenrod sel "raiseTool false" prevents 'define' from trying to bring up the Axes/ Planes/Centroids tool (and update that tool's table) for every centroid created. The other options control the size and color of the centroid. I had the small ligand selected so 'sel' is my atom spec. I tried it a second time with ":35" (which also selects the small ligand) and that worked too. FYI, "~define" will get rid of all the centroids. I hope this helps somehow. Are you sure you had the "Interpret script as" part of the per-frame dialog set to "Chimera commands"? I can't imagine that you didn't, since you would have gotten some kind of error if it had been interpreted as Python. --Eric Eric Pettersen UCSF Computer Graphics Lab http://www.cgl.ucsf.edu
{kind=link}