
Dear Elaine Thank you for your time and kind response. I apologize if this is outside of our business hours due to the time difference. Thank you also for your comments regarding hydrogen bonding. I am afraid that I have not studied enough myself, but I would like to confirm one point. I have seen 3.5Å and 3.7Å as the distance of hydrogen bonding in many papers, but am I correct in understanding that the distance between two atoms is 4Å or less as a reasonable distance for hydrogen bonding to be formed? Thank you for attaching the diagram regarding the hydrophobic interaction. I will check it out by actually operating the system myself. --------------------------------------------------------------------------- 松井 健治 東京農工大学大学院 工学府産業技術専攻 修士 2年 〒184-8588 東京都小金井市中町2-24-16 Mail: s214903z@st.go.tuat.ac.jp Kenji Matsui Graduate School of Tokyo University of Agriculture and Technology M2 2-24-16, Nakamachi, Koganei-shi, Tokyo 184-8588, Japan Mail: s214903z@st.go.tuat.ac.jp 2022年6月18日(土) 1:06 Elaine Meng <meng@cgl.ucsf.edu>:
Hi Kenji, "Examine residues" just means to look at the residues in the Chimera graphics window, and put the mouse cursor over them to see a pop-up label of residue number, etc.
However, these menu actions that you mentioned: (1) Select... Residue... Amino acid category... hydrophobic (2) Actions... Atoms/Bonds... Show ... do not specifically identify hydrophobic interactions with your ligand. Instead they would show ALL of the hydrophobic amino acids in your whole structure, no matter where they are.
There is no tool to identify only hydrophobic interactions. Instead you have to look at the residues near the ligand yourself and see which parts of the pocket are hydrophobic. There are tools that might help do part of this, however:
(A) Find Contacts tool. You can use this to find ALL contacts with ligand, but it does not tell you which contacts are polar or which are hydrophobic. This tutorial gives an example of using Find Contacts to identify contacts with ligand named FPS <https://www.rbvi.ucsf.edu/chimera/docs/UsersGuide/tutorials/squalene.html
(B) You can show the surface of the protein and color it by hydrophobicity (e.g. menu: Presets... interactive 3, there is an example below) ... the orange parts are the hydrophobic amino acids. However, this does not mean it is a hydrophobic interaction with your ligand, you still have to see whether the ligand has hydrophobic atoms and where they are in the pocket, whether or not they are near the orange surfaces.
Your session is very confusing because it has a target (#0) and many docked ligands (#1) plus three different combination models (#2-4 but only #2 is shown). If you are trying to analyze #2, probably better to close all that other stuff first (close #0,1,3,4) as long as you already have them saved as a session file. You can make a new session with only the part that you are trying to analyze.
Example commands to show receptor atoms within 5 Angstroms of ligand residues named LIG, except hiding the hydrogens on cartons (atom type HC): close #0,1,3,4 ~ribbon ~select repr stick show :LIG z<5 color byhet ~disp HC
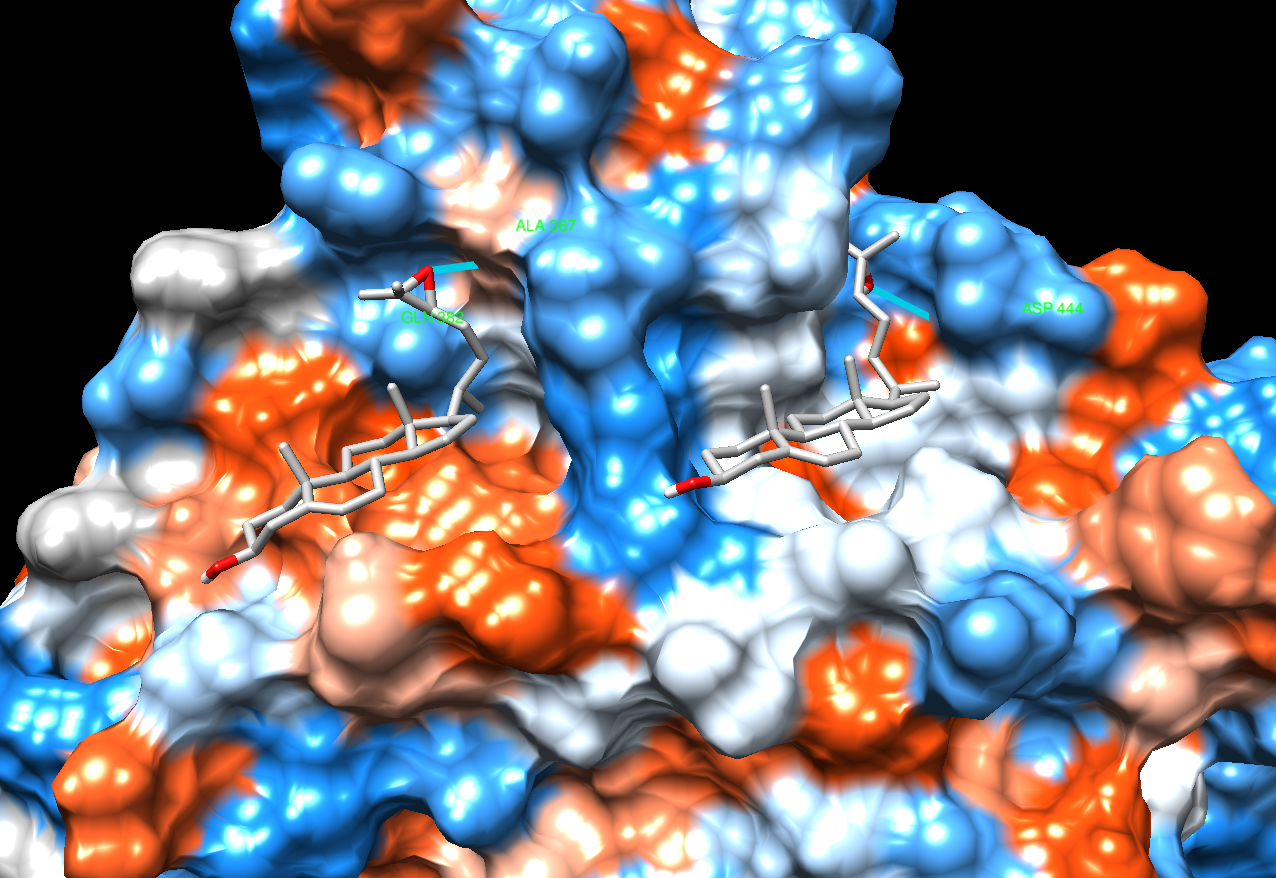
See image. I can see you have two lines drawn to show H-bonds, I guess, but they do not look like good H-bonds. In fact, if you put the mouse over the one on the right (in Chimera, not the image), you can see that it is 4.9 Angstroms long, which is way too long for a reasonable H-bond.
Also your combination model seems to be messed up somehow because it thinks the ligands are part of the protein and get put inside the surface unless I use some extra commands when trying to show receptor surface hydrophobicity.
command: surfcat rec protein menu: Presets... interactive 3 (hydrophobicity surface) command: surf rec command: ~surf :lig
See image. The pocket on the left is mostly hydrophobic (orange) but the one on the right is not. Also it does not look like the ligands fit the pocket shapes very well.
I hope this helps, Elaine ----- Elaine C. Meng, Ph.D. UCSF Chimera(X) team Department of Pharmaceutical Chemistry University of California, San Francisco
On Jun 16, 2022, at 11:27 PM, Kenji MATSUI via Chimera-users < chimera-users@cgl.ucsf.edu> wrote:
Dear Chimera
Thank you for taking the time to answer my question the other day.
I also apologize for contacting you outside of business hours due to the time difference.
I'm Kenji Matsui, a second-year master's student at a national university in Japan.
I have shown hydrophobic interactions between ligands and amino acid residues, how should I display them?
I refer to these methods.
But, I did not know where to find Examine residues.
Therefore, could you please tell me the location of Examine residues?
If possible, we would appreciate it if you could attach a diagram after the operation so that we can confirm the operating procedure.
A:Find hydrophobic interactions Select >> Residue(s) (either in entire structure or within a selected set (see above $ or %)) >> Amino acid category >> hydrophobic >> Action >> Atoms/Bonds >> Show >> Examine residues >> Mouse over atoms or Left click on them to identify them --------------------------------------------------------------------------- 松井 健治 東京農工大学大学院 工学府産業技術専攻 修士 2年 〒184-8588 東京都小金井市中町2-24-16
Kenji Matsui Graduate School of Tokyo University of Agriculture and Technology M2 2-24-16, Nakamachi, Koganei-shi, Tokyo 184-8588, Japan Mail: s214903z@st.go.tuat.ac.jp <LXRα_24,25_epoxy_hydrogen_1.85_1.135.py>
{kind=link}
{kind=link}