
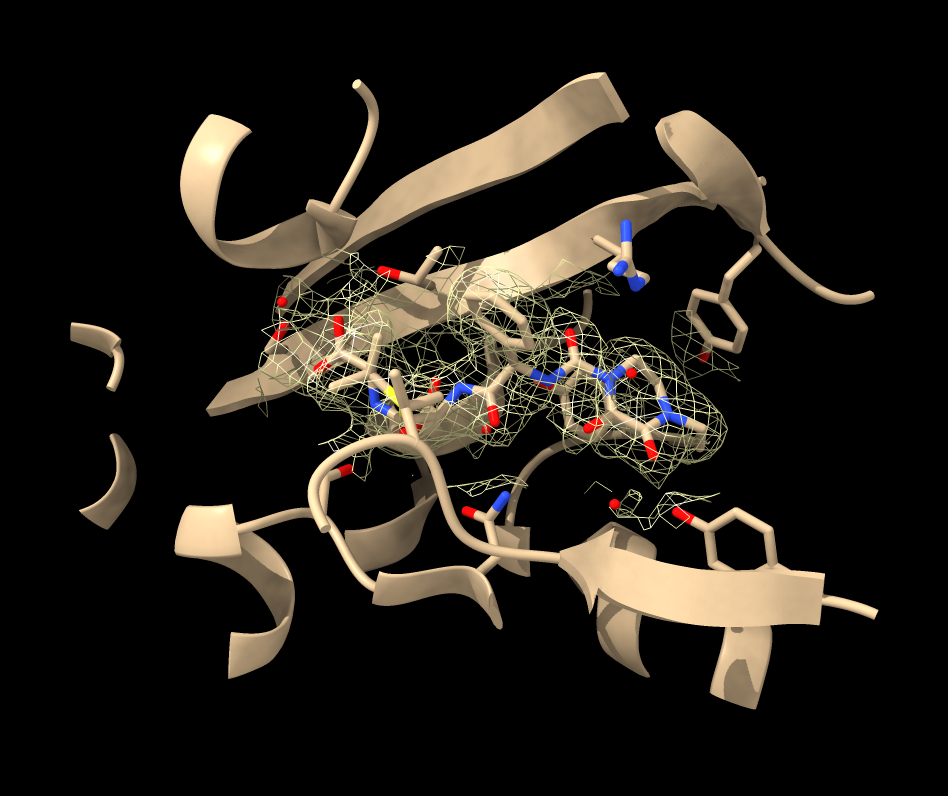
Hi Kelvin, Sometimes the PDB model of an X-ray structure lies outside the crystal unit cell map provide by the EDS and that is what you see with 4kqo. To get the map to cover the atomic model by using crystall symmetry use ChimeraX command volume cover open 4kqo open 4kqo from eds volume cover #2 atomBox #1 That creates map #3 that covers the atomic model. Then it is useful to hide the density except near the piperacillin ligand (residue JPP) in chain A using volume zone # 3 near /A:JPP Then I lower the threshold of the map volume #3 level 0.09 To get a better view of the density near the ligand you can also hide the atomic structure except near the ligand by first selecting the part near the ligand (within 8 Angstroms) sel /A:JPP :< 8 then hiding the part not selected hide ~sel atoms,ribbon There are ways to do these things with toolbar buttons and menus but it is easier for me to tell you the commands. Tom 
On Jul 12, 2024, at 9:33 AM, Elaine Meng via ChimeraX-users <chimerax-users@cgl.ucsf.edu> wrote:
Hi Kelvin, First make sure of which program you are using. You sent this to the chimera-users mailing list which is for the program Chimera. However, the commands that you mention are for ChimeraX, so you must really be using chimeraX instead. In the future, please use chimerax-users@cgl.ucsf.edu <mailto:chimerax-users@cgl.ucsf.edu> for ChimeraX questions (CC'd here). I included both since the answer may involve both.
Everything below is for ChimeraX unless stated otherwise:
(1) Is this substrate included in the experimental structure? If the experiment was not done with the substrate bound, there is no reason to expect the density map to include density for it. 4kqo does not have a density map in EDS, so I guess you meant 4kq0. 4kq0 has plenty of density near the ligand residue MED, if that's what you meant by "substrate." You just need to use the slider in the Volume Viewer to change the isosurface level of the density ("volume" command below "level" option, but for interactive use, it is easier to use the slider in the GUI). To make it easier to view you can also limit the density to a zone near that ligand with the command "volume zone"
open 1kq0 open 1kq0 from eds view ligand volume #2 level 0.01339 volume zone #2 near :MED
The isosurface level in the example above is lower than what is shown initially when you open the map. However, I have no opinion as to what value exactly you should use. See help for "volume" and "Volume Viewer"
<https://rbvi.ucsf.edu/chimerax/docs/user/commands/volume.html#general> <https://rbvi.ucsf.edu/chimerax/docs/user/commands/volume.html#zone> <https://rbvi.ucsf.edu/chimerax/docs/user/tools/volumeviewer.html>
(2) if you use "Join Models" section of "Build Structure" to create the bond (before combining) it will move one model to use your specified input bond distance and angles, and automatically combine the models. <https://rbvi.ucsf.edu/chimerax/docs/user/tools/buildstructure.html>
If you combine first and then add a bond, it won't move the atoms and the bond distance and angles may be poor, but it depends on your situation as to which approach is better, i.e. I'm not saying the way you did it was wrong. You may also be interested in using the other sections in Build Structure (see link above): Adjust Bonds, Adjust Torsions
You can also measure and change angles with "angle" and "torsion" commands. You can also rotate bonds manually using the "rotate bond" mousemode. <https://rbvi.ucsf.edu/chimerax/docs/user/commands/torsion.html> <https://rbvi.ucsf.edu/chimerax/docs/user/commands/angle.html> <https://rbvi.ucsf.edu/chimerax/docs/user/commands/ui.html#mousemode>
(3) there isn't a simple minimization tool that handles nonstandard residues in ChimeraX yet. You could save a PDB file of the result from the above and use Chimera instead, which has a Minimize Structure tool.
I hope this helps, Elaine ----- Elaine C. Meng, Ph.D. UCSF Chimera(X) team Resource for Biocomputing, Visualization, and Informatics Department of Pharmaceutical Chemistry University of California, San Francisco
On Jul 12, 2024, at 3:26 AM, Kelvin Chung via Chimera-users <chimera-users@cgl.ucsf.edu> wrote:
Good day I am a researcher studying antibiotic interaction and trying to make a model with a substrate covalently bound to the active site. There is a homolog with a crystal structure (PDB: 4KQO), and I am going to add the bound substrate in this pdb file to my target protein where the mode was generated by alphafold server.
Although the workflow shall be simple, I still have some problems, could you kindly help?
1. Firstly, I would like to check whether the model of 4KQO is good enough for modeling, by examining the electron density map (open 4KQO, open 4KQO from eds). However, I found that it is a bit different from the tutorial (https://www.cgl.ucsf.edu/chimerax/docs/quickstart/index.html, pdb:1a0m) that the electron density of 4KQO did not cover the substrate molecule. I am totally ignorant in crystallography, so, how could I visualize the electron density of the substrate? by any symmetry operation / command so that I could check whether there is "electron density in the substrate" ?
2. I aligned 4KQO with the alphafold generated model, and I deleted the protein from 4KQO, and combined the two model, make a bond between the active residue with the substrate, however, I am not sure how to (1) check the bond length and angle is appropriately assigned, and (2) do energy minimization to reorientate side change to eliminate the crash with the substrate.
Thank you very much for your help in advance
Have a nice weekend
_______________________________________________ ChimeraX-users mailing list -- chimerax-users@cgl.ucsf.edu <mailto:chimerax-users@cgl.ucsf.edu> To unsubscribe send an email to chimerax-users-leave@cgl.ucsf.edu <mailto:chimerax-users-leave@cgl.ucsf.edu> Archives: https://mail.cgl.ucsf.edu/mailman/archives/list/chimerax-users@cgl.ucsf.edu/
{kind=link}