
3 Apr
2018
3 Apr
'18
6:18 p.m.
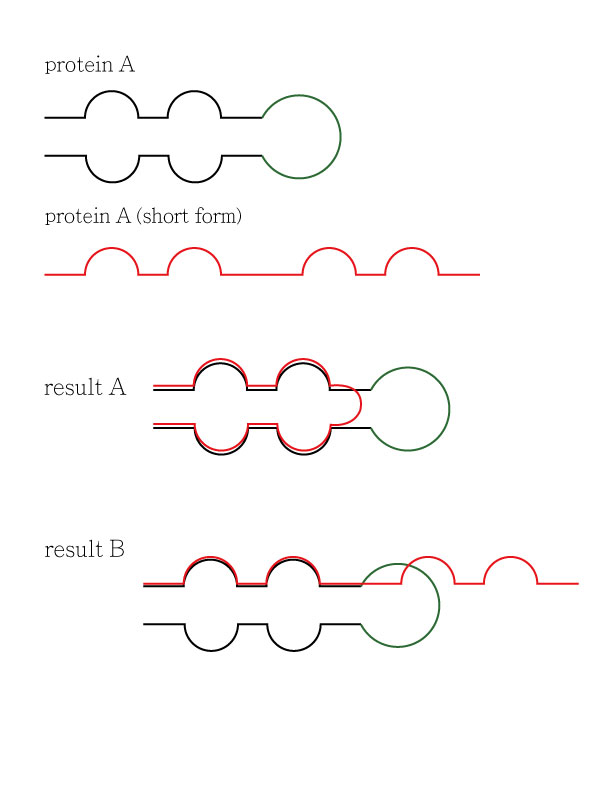
Hi my name is Teddy, I used UCSF Chimera for my research, and it is a very good software! I appreciate you made it. I have a question about structure comparison, I usually use Matchmaker to overlap 2 similar protein fragment to compare the difference. I wonder if the structure is bent or twisted during the matching process, in order to fit the reference structure more? For example, the attachment is for demonstration. There is a protein A, and protein A have a short from that miss some amino acid (the green part is missed). So the result produced by the Matchmaker would be A or B? If the result is A, can I make result B by UCSF Chimera? Thank you!
{kind=link}