
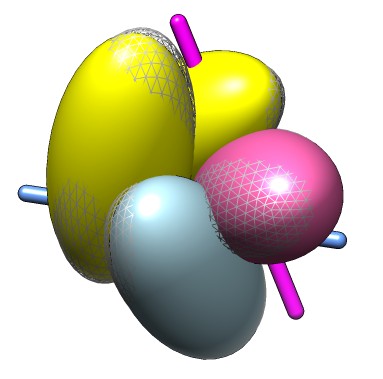
Hi Bobby, The place to send Chimera questions is the Chimera mailing list chimera-users at cgl.ucsf.edu. Here are some ideas about how to measure the rotation of one dimer of your homotetramer on ligand binding. Say the tetramer has chains A,B,C,D and we want to see how dimer CD has moved relative to dimer AB from unbound to bound PDB models. The idea is to use the Chimera "match" command with the showMatrix option. http://www.cgl.ucsf.edu/chimera/docs/UsersGuide/midas/match.html First step is that I want to create PDB files for bound and unbound states where the AB dimers are aligned. Suppose model #0 is unbound and model #1 is bound. This command will move the bound model AB to overlap the unbound model AB match #1:.A,.B #0:.A,.B Now align bound CD with unbound CD and report the rotation/translation with the showMatrix option: match showMatrix true #1:.C,.D #0:.C,.D The output in the Reply Log (Favorites menu) looks like the following. I used a dimer example instead of a tetramer to produce this output. Executing match ['#0:.B@CA', '#1:.A@CA'], no iteration Motion from original file coordinates Matrix rotation and translation -0.31727374 -0.31084290 0.89594311 19.18030808 -0.30378517 0.92828353 0.21448604 7.82438547 -0.89836070 -0.20412345 -0.38894945 20.67993251 Axis -0.22719634 0.97384144 0.00383052 Axis point 17.54082727 0.00000000 3.53442960 Rotation angle (degrees) 112.89041450 Shift along axis 3.34122976 Motion from last position Matrix rotation and translation -0.31727374 -0.31084290 0.89594311 19.18030808 -0.30378517 0.92828353 0.21448604 7.82438547 -0.89836070 -0.20412345 -0.38894945 20.67993251 Axis -0.22719634 0.97384144 0.00383052 Axis point 17.54082727 0.00000000 3.53442960 Rotation angle (degrees) 112.89041450 Shift along axis 3.34122976 RMSD between 16 atom pairs is 0.482 angstroms You would look at motion from last position, specifically the rotation angle. For your case (tetramers built from 1pfk and 2pfk) there is a problem that the exact same atoms are not in the two tetramers. The match command requires that you specify an exact pairing of atoms. You can use the Match Maker dialog or mmaker command which will figure out the atom pairing for you. But match maker doesn't report the motion so you would use the "measure rotation" command to do that. http://www.cgl.ucsf.edu/chimera/docs/UsersGuide/midas/mmaker.html http://www.cgl.ucsf.edu/chimera/docs/UsersGuide/midas/measure.html mmaker #1:.A,.B #0:.A,.B pair ss write relative #0 #1 /tmp/bound-ab-aligned.pdb close #1 open #1 /tmp/bound-ab-aligned.pdb mmaker #1:.C,.D #0:.C,.D pair ss measure rotation #1 #0 showSlabs true Here I aligned the AB dimer then saved the bound version to a file, closed the bound model, and opened the saved file. Did that close/open business because the measure rotation command is going to report values relative to the original file orientations. It doesn't report the motion from the last position because it doesn't remember what the last position was. The match and mmaker commands rotate the coordinate axes for one model and the "measure rotation" command reports the rotation between the coordinate axes of two models. Each model has its own coordinate axes which are initially aligned when the data is opened. I tried this with tetramers built from 1pfk and 2pfk dimers using the Chimera unit cell tool (2x2x2 cells) and the "combine" command and displayed the results with ellipsoids using the "measure intertia" command with the perChain true option. I've attached an image that shows the AB dimer in yellow, the CD dimer in blue and pink, 1pfk in solid and 2pfk in mesh, with separate rotation axes for the C and D rotations which appear quite different and are 2 to 3 degrees. I encountered an unpleasant bug that you saw before with the Chimera morph conformations tools that mmaker did not match chains C with C and D with D, instead matching C with D and D with C. It seems there is currently no way to control this other than modifying the PDB files (renumbering atoms?). Changing the order of the C and D in ":.C,.D" did not help. I've filed (another) bug report for this. http://plato.cgl.ucsf.edu/trac/chimera/ticket/8501 Tom -------- Original Message -------- From: Bobby Laird To: Thomas Goddard Date: 6/2/10 8:38 AM
Hi Tom,
I was not sure if you would remember me so I attached the previous e-mails that you were kins enough to CC to me a few months ago. I was wondering if you could give me any advice. I am still attempting to compare the quaternary structure of a homotetramer (320 aa's for each monomer). More specifically it appears that when certain ligands are bound there is a rotation about one of the dimer-dimer interfaces. I used the axis function in chimera to approximate angles in order to get a degree of rotation and was wondering if you could give me a basic explanation of how the program calculates these angles? Also, I wanted to create a very simplified cartoon figure of each of the pdb's so that I can superimpose and emphasize the rotation that occurs. Do you have any idea how I could accomplish this? I have tried rendering space filled models and removing the lighting so that the figure appears 2-D but when I overlay the pdb's the textures become very obvious. Sorry for dumping all this on you but I wasn't quite sure where to go with these questions. Thank you very much for for your time.
Bobby Laird
{kind=link}