
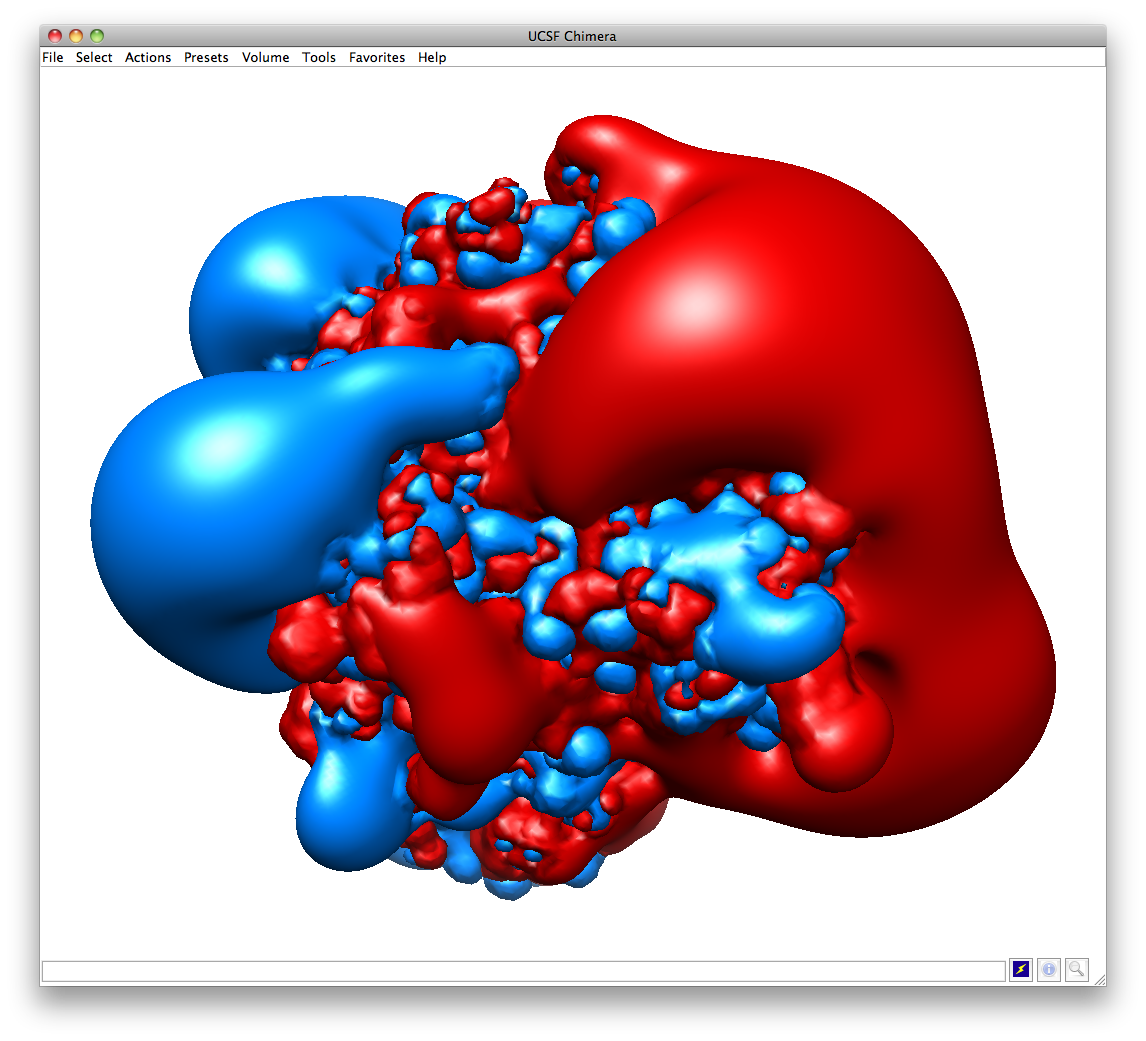
Hi Rebecca, You might also want to try showing the actual isosurfaces side-by-side. If the differences are dramatic, that might make a more impressive visual. Open your ".dx" files from APBS in Chimera and then use "Tools > Volume Data > Volume Viewer". Click on the histogram or change the "step" to display the isosurfaces. Adjust one threshold to have a "Level" of "-1" or maybe "-2" and color it blue. Then adjust the other threshold to have a "Level of "1" or maybe "2" and color it red. See attached for an example. My $0.02, Darrell NIAID Darrell Hurt, Ph.D. Section Head, Computational Biology Bioinformatics and Computational Biosciences Branch (BCBB) OCICB/OSMO/OD/NIAID/NIH 31 Center Drive, Room 3B62B, MSC 2135 Bethesda, MD 20892-2135 Office 301-402-0095 Mobile 301-758-3559 http://bioinformatics.niaid.nih.gov (Within NIH) http://exon.niaid.nih.gov (Public) Disclaimer: The information in this e-mail and any of its attachments is confidential and may contain sensitive information. It should not be used by anyone who is not the original intended recipient. If you have received this e-mail in error please inform the sender and delete it from your mailbox or any other storage devices. National Institute of Allergy and Infectious Diseases shall not accept liability for any statements made that are sender's own and not expressly made on behalf of the NIAID by one of its representatives. On 4/11/12 1:54 PM, "Rebecca Swett" <rswett@chem.wayne.edu> wrote:
That actually might work for me. The odd thing about the structures i'm comparing is that while the surface shape is nearly identical, the sequence identity is quite disparate. I'll play with it and let you know how it turns out. Thanks again for your suggestions. ~Rebecca
On 4/11/2012 1:41 PM, Elaine Meng wrote:
Hi Rebecca, I was also thinking you might try smoothing the difference map to see if that better brings out the major features. Various kinds of map smoothing (Gaussian filtering, etc.) can be done with "vop" command options or the Volume Filter tool (under Tools...Volume Data).
<http://www.cgl.ucsf.edu/chimera/docs/ContributedSoftware/volumeviewer/ga ussian.html>
Of course, the more processing you do, the more you have to explain to your audience!
I'm not sure what you are getting at with the residue charge issue. The APBS map doesn't have charges, it only has the potential resulting from those charges. You already know for the most part which residues are charged (Asp/Glu negative, Lys/Arg positive, His being the ambiguous case) and that only involves the structure, not the map.
While you could use the Values at Atom Positions tool to get ESP values mapped to atom positions,
<http://www.cgl.ucsf.edu/chimera/docs/ContributedSoftware/density/density .html>
...and sum over atom values to get the residue values with Attribute Calculator,
<http://www.cgl.ucsf.edu/chimera/docs/ContributedSoftware/calculator/calc ulator.html>
...that doesn't seem particularly useful applied to the same atoms that gave rise to the potential. Typically it would be used to map potential from one molecule (say a receptor) onto other molecules not used to calculate the potential (say different small molecule ligands in the binding site).
Maybe I misunderstood the question, though. Best, Elaine
On Apr 11, 2012, at 10:18 AM, Rebecca Swett wrote:
Thanks for the quick reply. I have the side by sides. I'll see if I get anything particularly wonky if I try the subtract. Alternatively, can you think of a way I could output per-residue charge from an APBS map? I might be able to do a simple subtract and render by attribute to get a rough approximation. ~Rebecca
_______________________________________________ Chimera-users mailing list Chimera-users@cgl.ucsf.edu http://plato.cgl.ucsf.edu/mailman/listinfo/chimera-users
{kind=link}