How to predict/model the structure of two known structure proteins

Dear UCSF developers, I am Hoang, working in IPK in Germany. I am following your series online tutorials about how to use UCSF Chimera. This program is very useful for me. However, I did not see your video showing how to predict/model the structure of a fusion protein that is made by combination of two known structure proteins in PDB (for example gi|534286646|pdb|4KRN| and gi|390136394|pdb|4DDF|A). Is this function available in your software? If yes can you show me how to manipulate with it. I would like appreciate it very much Thank you for your attention and I am looking forward to hearing from you. Best regards Hoang Phan

Dear Hoang, Yes, but there are a few steps. You would need to: (1) use a text-editor to make a plain text fasta-format file of the fusion protein sequence, if you don’t already have such a file. Fasta format description: <http://blast.ncbi.nlm.nih.gov/blastcgihelp.shtml> Make the name of your file end with “.fasta" (2) open the fasta file in Chimera. Then from sequence window menu: Info… Blast Protein to search the PDB for matching structures. In the Blast Protein results, find the two structure proteins and choose both lines (click, ctrl-click in the results dialog), then click the “Show in MAV” and “Load Structure” buttons at the bottom of the dialog. <http://www.rbvi.ucsf.edu/chimera/docs/UsersGuide/blast.html> (3) now you will have a new sequence alignment window with 3 sequences in it: the fusion protein and the two protein structures, and in the main Chimera window, the two structures. (4) In Chimera, delete any extra protein chains. In other words, if there are extra copies of those structures, just delete them so that you have only one copy to use as the template. Make sure that the remaining copy of each is associated with its sequence in the alignment (sequence alignment window menu: Structures… Associations…) <http://www.rbvi.ucsf.edu/chimera/docs/ContributedSoftware/multalignviewer/fr...> <http://www.rbvi.ucsf.edu/chimera/docs/ContributedSoftware/multalignviewer/mu...> (5) position the two structures so that the termini are in a somewhat reasonable place relative to each other to template the fusion protein. You can “freeze” one in place by deactivating it and move just the other with the mouse as described here: <http://www.rbvi.ucsf.edu/chimera/docs/UsersGuide/mouse.html#activedef> 6) from the sequence alignment window menu choose: Structure… Modeller (homology) to show the Modeller dialog. Choose the query as the target and both structures as the template, etc. as in the modeling tutorials. You may also want to turn on “Use thorough optimization” in the Advanced Options section. I don’t know which homology-modeling tutorial you have been using, but here is one: <http://www.rbvi.ucsf.edu/chimera/docs/UsersGuide/tutorials/dor.html> <http://www.rbvi.ucsf.edu/chimera/docs/UsersGuide/tutorials/dor.html#modeller> I hope this helps, Elaine ----- Elaine C. Meng, Ph.D. UCSF Computer Graphics Lab (Chimera team) and Babbitt Lab Department of Pharmaceutical Chemistry University of California, San Francisco On Jan 21, 2015, at 2:16 AM, Phan Trong Hoang <hoang@ipk-gatersleben.de> wrote:
Dear UCSF developers, I am Hoang, working in IPK in Germany. I am following your series online tutorials about how to use UCSF Chimera. This program is very useful for me. However, I did not see your video showing how to predict/model the structure of a fusion protein that is made by combination of two known structure proteins in PDB (for example gi|534286646|pdb|4KRN| and gi|390136394|pdb|4DDF|A). Is this function available in your software? If yes can you show me how to manipulate with it. I would like appreciate it very much Thank you for your attention and I am looking forward to hearing from you. Best regards Hoang Phan
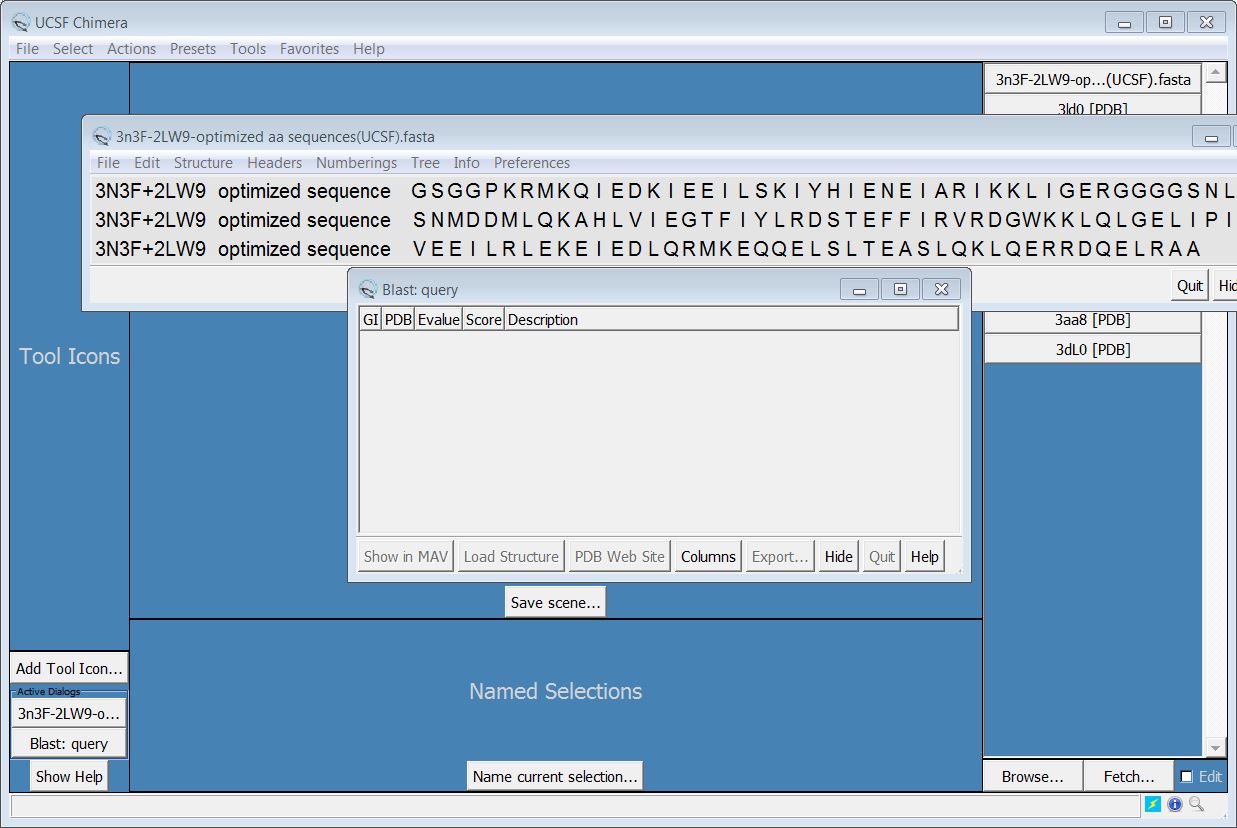
Dear Meng, Thank you very much for your reply. I have tried as you wrote (please see figure 1), but I face the problem. There is a Blast:query appearance but there is no information about PDB....please have a look at the attached picture (figure 2). I tried with the sequence of one protein in PDB. The same table appeared with no more information. Could you show me how to fix it or there are other steps to avoid this step. Thank you Best regards Hoang -----Original Message----- From: Elaine Meng [mailto:meng@cgl.ucsf.edu] Sent: Mittwoch, 21. Januar 2015 23:02 To: Phan Trong Hoang Cc: chimera-users@cgl.ucsf.edu Subject: Re: [Chimera-users] How to predict/model the structure of two known structure proteins Dear Hoang, Yes, but there are a few steps. You would need to: (1) use a text-editor to make a plain text fasta-format file of the fusion protein sequence, if you don't already have such a file. Fasta format description: <http://blast.ncbi.nlm.nih.gov/blastcgihelp.shtml> Make the name of your file end with ".fasta" (2) open the fasta file in Chimera. Then from sequence window menu: Info... Blast Protein to search the PDB for matching structures. In the Blast Protein results, find the two structure proteins and choose both lines (click, ctrl-click in the results dialog), then click the "Show in MAV" and "Load Structure" buttons at the bottom of the dialog. <http://www.rbvi.ucsf.edu/chimera/docs/UsersGuide/blast.html> (3) now you will have a new sequence alignment window with 3 sequences in it: the fusion protein and the two protein structures, and in the main Chimera window, the two structures. (4) In Chimera, delete any extra protein chains. In other words, if there are extra copies of those structures, just delete them so that you have only one copy to use as the template. Make sure that the remaining copy of each is associated with its sequence in the alignment (sequence alignment window menu: Structures... Associations...) <http://www.rbvi.ucsf.edu/chimera/docs/ContributedSoftware/multalignviewer/fr...> <http://www.rbvi.ucsf.edu/chimera/docs/ContributedSoftware/multalignviewer/mu...> (5) position the two structures so that the termini are in a somewhat reasonable place relative to each other to template the fusion protein. You can "freeze" one in place by deactivating it and move just the other with the mouse as described here: <http://www.rbvi.ucsf.edu/chimera/docs/UsersGuide/mouse.html#activedef> 6) from the sequence alignment window menu choose: Structure... Modeller (homology) to show the Modeller dialog. Choose the query as the target and both structures as the template, etc. as in the modeling tutorials. You may also want to turn on "Use thorough optimization" in the Advanced Options section. I don't know which homology-modeling tutorial you have been using, but here is one: <http://www.rbvi.ucsf.edu/chimera/docs/UsersGuide/tutorials/dor.html> <http://www.rbvi.ucsf.edu/chimera/docs/UsersGuide/tutorials/dor.html#modeller> I hope this helps, Elaine ----- Elaine C. Meng, Ph.D. UCSF Computer Graphics Lab (Chimera team) and Babbitt Lab Department of Pharmaceutical Chemistry University of California, San Francisco On Jan 21, 2015, at 2:16 AM, Phan Trong Hoang <hoang@ipk-gatersleben.de> wrote:
Dear UCSF developers, I am Hoang, working in IPK in Germany. I am following your series online tutorials about how to use UCSF Chimera. This program is very useful for me. However, I did not see your video showing how to predict/model the structure of a fusion protein that is made by combination of two known structure proteins in PDB (for example gi|534286646|pdb|4KRN| and gi|390136394|pdb|4DDF|A). Is this function available in your software? If yes can you show me how to manipulate with it. I would like appreciate it very much Thank you for your attention and I am looking forward to hearing from you. Best regards Hoang Phan
{kind=link}
{kind=link}
Dear Hoang, Blast should find the matching PDB structures with those default settings. Just now I tried using a fasta file with only the last line of sequence shown in your image, and it correctly found PDB 2LW9. The results dialog first appears empty, but later when the search is done the results appear. So maybe you just needed to wait a few minutes for the results. However, if you wait a long time and still don't get results, you could skip the Blast part and instead: (A) create (outside of Chimera) a 3-sequence alignment containing your target fusion protein and the sequences of each of the template PDB structures, in any of the formats Chimera can read: <http://www.rbvi.ucsf.edu/chimera/docs/UsersGuide/filetypes.html#alignment> (B) open that sequence alignment and the two template PDB structures in Chimera, then proceed starting with step #4 in my previous message. <http://plato.cgl.ucsf.edu/pipermail/chimera-users/2015-January/010645.html> Another possibility that I forgot to mention before is to use the new "mda" multidomain assembler command (in Chimera 1.10.1 or newer daily build) with your fusion-protein fasta file as input. This automates several of the steps in my previous message, including running Blast, up to the point of opening the Modeller dialog. Of course, Blast will have to be working correctly, but my tests suggest that it is. The "mda" command has several options, and I don't know whether you would need to use any of those options for it to find the desired template structures. However, it might still be worth a try because then you don't have to do steps #2-5 yourself. See the syntax and option information here: <http://www.rbvi.ucsf.edu/chimera/docs/UsersGuide/midas/mda.html> I hope this helps, Elaine On Jan 22, 2015, at 6:05 AM, Phan Trong Hoang <hoang@ipk-gatersleben.de> wrote:
Dear Meng, Thank you very much for your reply. I have tried as you wrote (please see figure 1), but I face the problem. There is a Blast:query appearance but there is no information about PDB....please have a look at the attached picture (figure 2). I tried with the sequence of one protein in PDB. The same table appeared with no more information. Could you show me how to fix it or there are other steps to avoid this step. Thank you Best regards Hoang
participants (2)
-
 Elaine Meng
Elaine Meng -
 Phan Trong Hoang
Phan Trong Hoang