
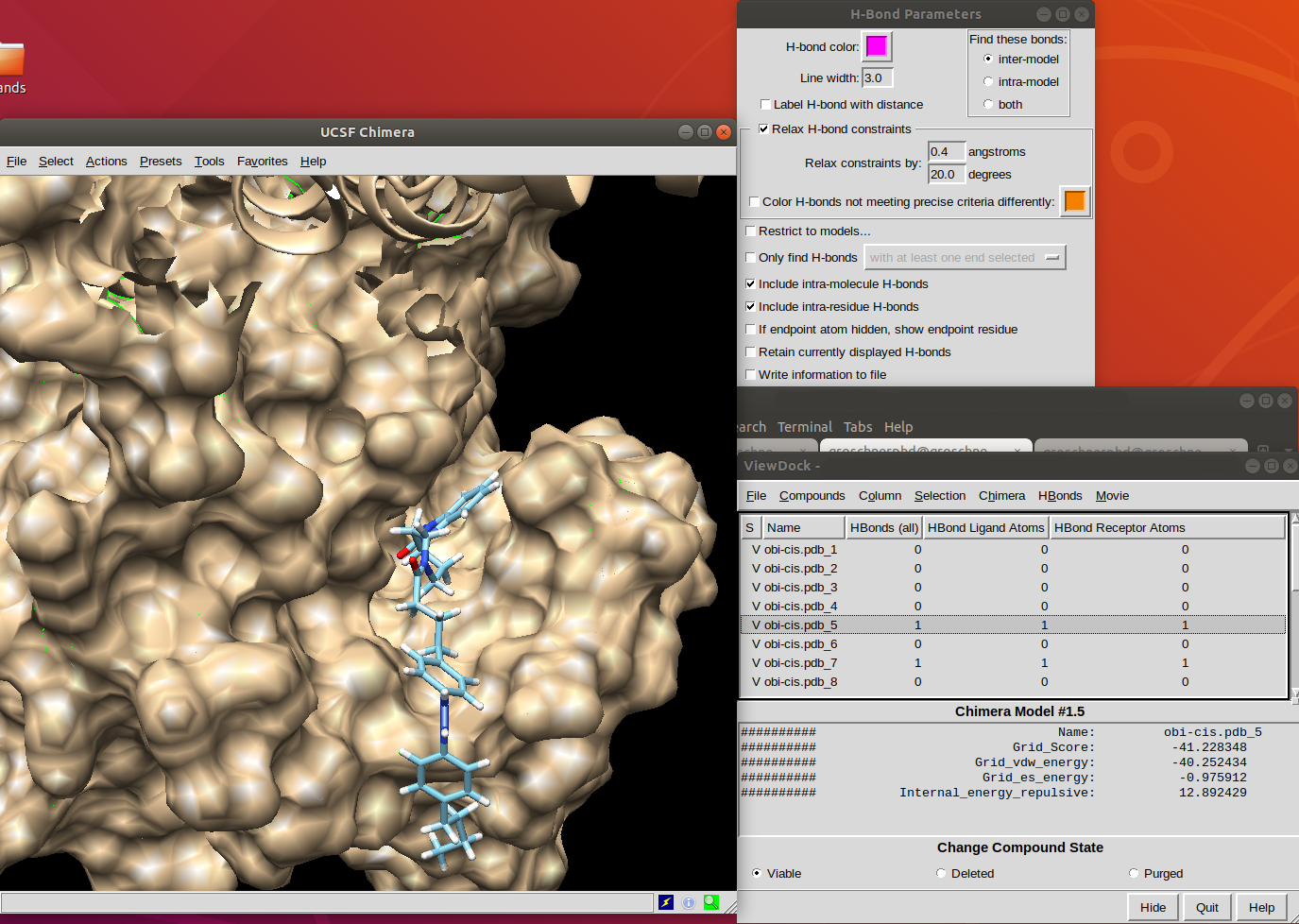
Hello, Does anybody know why the viewdock calculates Hbonds, but doesn't want to show them (in some cases-picture, not always)? Kind regards, Denis
{kind=link}

Hi Denis, It's a little hard to see when you have the molecular surface also shown, but I'm guessing you need to turn on the H-Bond option "If endpoint atom hidden, show endpoint residue" <http://www.rbvi.ucsf.edu/chimera/docs/ContributedSoftware/findhbond/findhbon...> This is because the H-bond line can't be shown unless the atoms on both of its ends are shown. That option will show more residues of the receptor so that all of the identified H-bonds will be shown. Regards, Elaine ----- Elaine C. Meng, Ph.D. UCSF Chimera(X) team Department of Pharmaceutical Chemistry University of California, San Francisco
On Jul 30, 2020, at 5:20 AM, Krivic, Denis <denis.krivic@medunigraz.at> wrote:
Hello, Does anybody know why the viewdock calculates Hbonds, but doesn't want to show them (in some cases-picture, not always)? Kind regards, Denis
participants (2)
-
 Elaine Meng
Elaine Meng -
 Krivic, Denis
Krivic, Denis