disulfide bonds ?

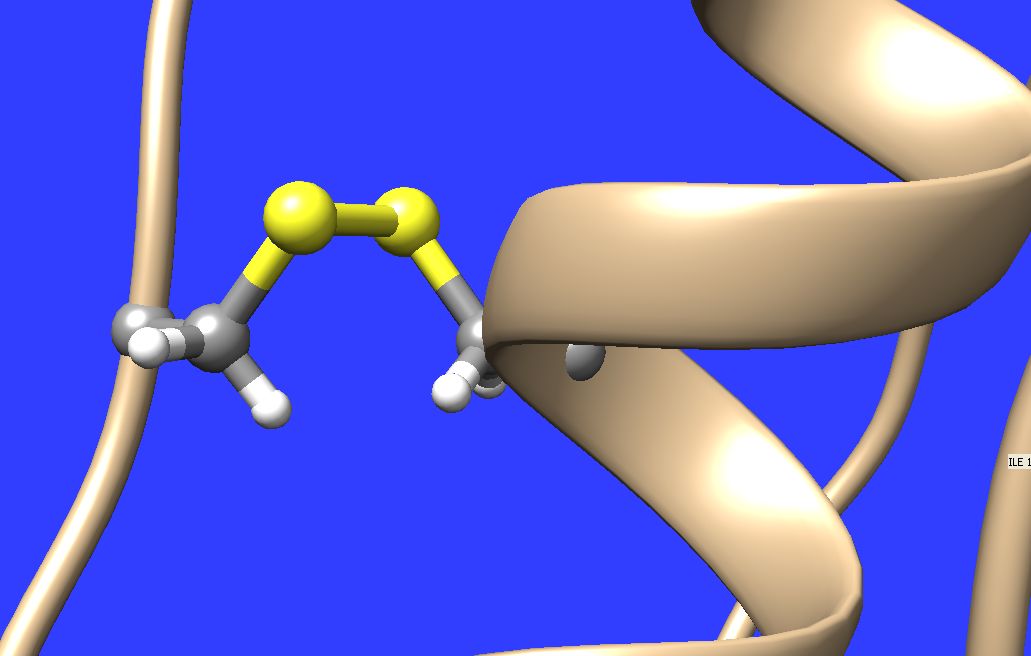
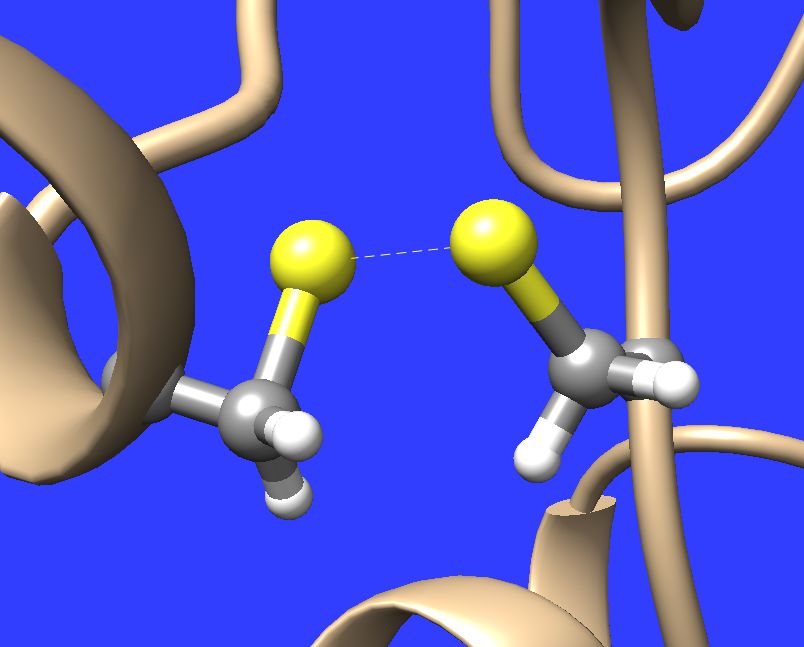
Hellow, I just visualized my Amber MD trajectory with protein containing several disulfide bonds. These bonds as well as all the other bonds are written in the given Amber topology (*.prmtop) file, which is read in by Chimera. Moreover the distance of sulfur atoms connected by disulfide bond is in all cases quite reasonable ( 2-2.1 A). In spite these facts Chimera draws some of those bonds in "stick" and some of them in "dashed line" style, even if one sets globally "stick" or "ball & stick" representation. Please see the attached illustrations. Why Chimera is doing such unwanted differences in graphical representations here ? It is possible, to force Chimera to draw all of those bonds unanimously in "stick" representation ? Thank you in advance for your help, Best wishes, Marek Maly -- Tato zpráva byla vytvořena převratným poštovním klientem Opery: http://www.opera.com/mail/
{kind=link}
{kind=link}

On Jul 14, 2014, at 8:57 AM, "Marek Maly" <marek.maly@ujep.cz> wrote:
Hellow,
I just visualized my Amber MD trajectory with protein containing several disulfide bonds. These bonds as well as all the other bonds are written in the given Amber topology (*.prmtop) file, which is read in by Chimera. Moreover the distance of sulfur atoms connected by disulfide bond is in all cases quite reasonable ( 2-2.1 A). In spite these facts Chimera draws some of those bonds in "stick" and some of them in "dashed line" style, even if one sets globally "stick" or "ball & stick" representation. Please see the attached illustrations.
Why Chimera is doing such unwanted differences in graphical representations here ?
It is possible, to force Chimera to draw all of those bonds unanimously in "stick" representation ?
Hi Marek, My best guess is that Chimera is mistakenly converting some of the the disulphide bonds into "missing structure" pseudobonds, which are supposed to be used to represent parts of a chain that missing because is couldn't be located in the experimental data -- which obviously is not the case here! The workaround to make those pseudobonds be depicted as stick is to open the Pseudobond Panel (in Tools/General Controls) and the double click on the "missing structure" group to bring up its attribute inspector. In that inspector, click the check box next to "Component Pseudobond Attributes" to reveal those options and then change "bond style" from wire to stick. There is an equivalent command: "setattr pb drawMode 1". Obviously the better solution is to have Chimera not make this mistake in the first place. As far as I know it shouldn't be already. So, I would need your prmtop file and at least a subset of your coordinate file in order to investigate. Also, what version of Chimera are you using? --Eric Eric Pettersen UCSF Computer Graphics Lab http://www.cgl.ucsf.edu
Hi Eric, thank you for the prompt and useful answer. The approach you suggested works ! But I already identified the source of the problem. In my Amber MD trajectory, there was also the first(starting) configuration of my protein (that from the *.inpcrd file). This starting configuration was not so precise as was obtained by the electron microscopy (not by X-ray or NMR technique). From this reason distances of some of the sulfur atoms which should be connected by disulfide bond was not reasonable in this starting configuration but higher like 3 A or 6 A. So in spite the fact that in the Amber topology (*.prmtop) file were clearly written all the disulfide bonds, Chimera evidently ignored those bonds in sulfur atom pairs where the distance was not in the reasonable range (close to 2A). Instead perhaps Chimera considered, that there are some missing parts or so and used "dashed line" instead "stick". So this was the first Chimera "mistake" which should not happen if she just simply accepted the full topology information from the "*.prmtop" file. Another problem was that even if in all consequent frames of the trajectory all the relevant sulfur atoms were in the reasonable distance (2-2.1 A - due to the fact that during the Amber simulation they were bonded), Chimera doesn't care about because she evaluate the overall structure just using the first frame of the trajectory. I would suggest here, that Chimera should not take her own decisions in such cases when molecular topology is fully defined in input files (e.g. Amber *.prmtop, mol2 etc.). Especially in case of the Amber *.prmtop file it is clear that structure should be OK, (e.g. without the missing parts) because otherwise this topology file would never be created. If Chimera find something strange in the structure (even if this structure was fully determined in the input file/s), she can always write the proper warnings, and that should be enough in such specific cases, I think. Best wishes, Marek Dne Mon, 14 Jul 2014 19:33:51 +0200 Eric Pettersen <pett@cgl.ucsf.edu> napsal/-a:
On Jul 14, 2014, at 8:57 AM, "Marek Maly" <marek.maly@ujep.cz> wrote:
Hellow,
I just visualized my Amber MD trajectory with protein containing several disulfide bonds. These bonds as well as all the other bonds are written in the given Amber topology (*.prmtop) file, which is read in by Chimera. Moreover the distance of sulfur atoms connected by disulfide bond is in all cases quite reasonable ( 2-2.1 A). In spite these facts Chimera draws some of those bonds in "stick" and some of them in "dashed line" style, even if one sets globally "stick" or "ball & stick" representation. Please see the attached illustrations.
Why Chimera is doing such unwanted differences in graphical representations here ?
It is possible, to force Chimera to draw all of those bonds unanimously in "stick" representation ?
Hi Marek, My best guess is that Chimera is mistakenly converting some of the the disulphide bonds into "missing structure" pseudobonds, which are supposed to be used to represent parts of a chain that missing because is couldn't be located in the experimental data -- which obviously is not the case here! The workaround to make those pseudobonds be depicted as stick is to open the Pseudobond Panel (in Tools/General Controls) and the double click on the "missing structure" group to bring up its attribute inspector. In that inspector, click the check box next to "Component Pseudobond Attributes" to reveal those options and then change "bond style" from wire to stick. There is an equivalent command: "setattr pb drawMode 1". Obviously the better solution is to have Chimera not make this mistake in the first place. As far as I know it shouldn't be already. So, I would need your prmtop file and at least a subset of your coordinate file in order to investigate. Also, what version of Chimera are you using?
--Eric
Eric Pettersen UCSF Computer Graphics Lab http://www.cgl.ucsf.edu
__________ Informace od ESET NOD32 Antivirus, verze databaze 10094 (20140714) __________
Tuto zpravu proveril ESET NOD32 Antivirus.
-- Tato zpráva byla vytvořena převratným poštovním klientem Opery: http://www.opera.com/mail/
Hi Marek, I'm glad my suggestion worked. I agree with you assessment, which is why I was hoping you could provide data files for testing. Nonetheless, I have gone ahead and made changes such that for formats where explicit topology files are provided, such as Amber, Chimera "trusts" the connectivity and does not change excessively long bonds into pseudobonds. Those changes will be in tonight's daily build and I'm hoping you can test them and let me know if they work. Look for daily builds dated July 15th or later. --Eric On Jul 15, 2014, at 4:08 AM, "Marek Maly" <marek.maly@ujep.cz> wrote:
Hi Eric, thank you for the prompt and useful answer.
The approach you suggested works !
But I already identified the source of the problem.
In my Amber MD trajectory, there was also the first(starting) configuration of my protein (that from the *.inpcrd file).
This starting configuration was not so precise as was obtained by the electron microscopy (not by X-ray or NMR technique).
From this reason distances of some of the sulfur atoms which should be connected by disulfide bond was not reasonable in this starting configuration but higher like 3 A or 6 A.
So in spite the fact that in the Amber topology (*.prmtop) file were clearly written all the disulfide bonds, Chimera evidently ignored those bonds in sulfur atom pairs where the distance was not in the reasonable range (close to 2A). Instead perhaps Chimera considered, that there are some missing parts or so and used "dashed line" instead "stick". So this was the first Chimera "mistake" which should not happen if she just simply accepted the full topology information from the "*.prmtop" file.
Another problem was that even if in all consequent frames of the trajectory all the relevant sulfur atoms were in the reasonable distance (2-2.1 A - due to the fact that during the Amber simulation they were bonded), Chimera doesn't care about because she evaluate the overall structure just using the first frame of the trajectory.
I would suggest here, that Chimera should not take her own decisions in such cases when molecular topology is fully defined in input files (e.g. Amber *.prmtop, mol2 etc.).
Especially in case of the Amber *.prmtop file it is clear that structure should be OK, (e.g. without the missing parts) because otherwise this topology file would never be created.
If Chimera find something strange in the structure (even if this structure was fully determined in the input file/s), she can always write the proper warnings, and that should be enough in such specific cases, I think.
Best wishes,
Marek
Dne Mon, 14 Jul 2014 19:33:51 +0200 Eric Pettersen <pett@cgl.ucsf.edu> napsal/-a:
On Jul 14, 2014, at 8:57 AM, "Marek Maly" <marek.maly@ujep.cz> wrote:
Hellow,
I just visualized my Amber MD trajectory with protein containing several disulfide bonds. These bonds as well as all the other bonds are written in the given Amber topology (*.prmtop) file, which is read in by Chimera. Moreover the distance of sulfur atoms connected by disulfide bond is in all cases quite reasonable ( 2-2.1 A). In spite these facts Chimera draws some of those bonds in "stick" and some of them in "dashed line" style, even if one sets globally "stick" or "ball & stick" representation. Please see the attached illustrations.
Why Chimera is doing such unwanted differences in graphical representations here ?
It is possible, to force Chimera to draw all of those bonds unanimously in "stick" representation ?
Hi Marek, My best guess is that Chimera is mistakenly converting some of the the disulphide bonds into "missing structure" pseudobonds, which are supposed to be used to represent parts of a chain that missing because is couldn't be located in the experimental data -- which obviously is not the case here! The workaround to make those pseudobonds be depicted as stick is to open the Pseudobond Panel (in Tools/General Controls) and the double click on the "missing structure" group to bring up its attribute inspector. In that inspector, click the check box next to "Component Pseudobond Attributes" to reveal those options and then change "bond style" from wire to stick. There is an equivalent command: "setattr pb drawMode 1". Obviously the better solution is to have Chimera not make this mistake in the first place. As far as I know it shouldn't be already. So, I would need your prmtop file and at least a subset of your coordinate file in order to investigate. Also, what version of Chimera are you using?
--Eric
Eric Pettersen UCSF Computer Graphics Lab http://www.cgl.ucsf.edu
__________ Informace od ESET NOD32 Antivirus, verze databaze 10094 (20140714) __________
Tuto zpravu proveril ESET NOD32 Antivirus.
-- Tato zpráva byla vytvořena převratným poštovním klientem Opery: http://www.opera.com/mail/
Hi Eric, thanks for your decisions. Sorry that I forget to send my files for testing. I was fully satisfied just with the complete explanation of this Chimera strange behavior :)) I will send you my input files off-line. Best wishes, Marek Dne Tue, 15 Jul 2014 20:42:39 +0200 Eric Pettersen <pett@cgl.ucsf.edu> napsal/-a:
Hi Marek, I'm glad my suggestion worked. I agree with you assessment, which is why I was hoping you could provide data files for testing. Nonetheless, I have gone ahead and made changes such that for formats where explicit topology files are provided, such as Amber, Chimera "trusts" the connectivity and does not change excessively long bonds into pseudobonds. Those changes will be in tonight's daily build and I'm hoping you can test them and let me know if they work. Look for daily builds dated July 15th or later.
--Eric
On Jul 15, 2014, at 4:08 AM, "Marek Maly" <marek.maly@ujep.cz> wrote:
Hi Eric, thank you for the prompt and useful answer.
The approach you suggested works !
But I already identified the source of the problem.
In my Amber MD trajectory, there was also the first(starting) configuration of my protein (that from the *.inpcrd file).
This starting configuration was not so precise as was obtained by the electron microscopy (not by X-ray or NMR technique).
From this reason distances of some of the sulfur atoms which should be connected by disulfide bond was not reasonable in this starting configuration but higher like 3 A or 6 A.
So in spite the fact that in the Amber topology (*.prmtop) file were clearly written all the disulfide bonds, Chimera evidently ignored those bonds in sulfur atom pairs where the distance was not in the reasonable range (close to 2A). Instead perhaps Chimera considered, that there are some missing parts or so and used "dashed line" instead "stick". So this was the first Chimera "mistake" which should not happen if she just simply accepted the full topology information from the "*.prmtop" file.
Another problem was that even if in all consequent frames of the trajectory all the relevant sulfur atoms were in the reasonable distance (2-2.1 A - due to the fact that during the Amber simulation they were bonded), Chimera doesn't care about because she evaluate the overall structure just using the first frame of the trajectory.
I would suggest here, that Chimera should not take her own decisions in such cases when molecular topology is fully defined in input files (e.g. Amber *.prmtop, mol2 etc.).
Especially in case of the Amber *.prmtop file it is clear that structure should be OK, (e.g. without the missing parts) because otherwise this topology file would never be created.
If Chimera find something strange in the structure (even if this structure was fully determined in the input file/s), she can always write the proper warnings, and that should be enough in such specific cases, I think.
Best wishes,
Marek
Dne Mon, 14 Jul 2014 19:33:51 +0200 Eric Pettersen <pett@cgl.ucsf.edu> napsal/-a:
On Jul 14, 2014, at 8:57 AM, "Marek Maly" <marek.maly@ujep.cz> wrote:
Hellow,
I just visualized my Amber MD trajectory with protein containing several disulfide bonds. These bonds as well as all the other bonds are written in the given Amber topology (*.prmtop) file, which is read in by Chimera. Moreover the distance of sulfur atoms connected by disulfide bond is in all cases quite reasonable ( 2-2.1 A). In spite these facts Chimera draws some of those bonds in "stick" and some of them in "dashed line" style, even if one sets globally "stick" or "ball & stick" representation. Please see the attached illustrations.
Why Chimera is doing such unwanted differences in graphical representations here ?
It is possible, to force Chimera to draw all of those bonds unanimously in "stick" representation ?
Hi Marek, My best guess is that Chimera is mistakenly converting some of the the disulphide bonds into "missing structure" pseudobonds, which are supposed to be used to represent parts of a chain that missing because is couldn't be located in the experimental data -- which obviously is not the case here! The workaround to make those pseudobonds be depicted as stick is to open the Pseudobond Panel (in Tools/General Controls) and the double click on the "missing structure" group to bring up its attribute inspector. In that inspector, click the check box next to "Component Pseudobond Attributes" to reveal those options and then change "bond style" from wire to stick. There is an equivalent command: "setattr pb drawMode 1". Obviously the better solution is to have Chimera not make this mistake in the first place. As far as I know it shouldn't be already. So, I would need your prmtop file and at least a subset of your coordinate file in order to investigate. Also, what version of Chimera are you using?
--Eric
Eric Pettersen UCSF Computer Graphics Lab http://www.cgl.ucsf.edu
__________ Informace od ESET NOD32 Antivirus, verze databaze 10094 (20140714) __________
Tuto zpravu proveril ESET NOD32 Antivirus.
-- Tato zpráva byla vytvořena převratným poštovním klientem Opery: http://www.opera.com/mail/
__________ Informace od ESET NOD32 Antivirus, verze databaze 10101 (20140715) __________
Tuto zpravu proveril ESET NOD32 Antivirus.
-- Tato zpráva byla vytvořena převratným poštovním klientem Opery: http://www.opera.com/mail/
Thanks for the input files. I'm happy to report the problem is indeed fixed. --Eric On Jul 15, 2014, at 1:57 PM, "Marek Maly" <marek.maly@ujep.cz> wrote:
Hi Eric, thanks for your decisions.
Sorry that I forget to send my files for testing. I was fully satisfied just with the complete explanation of this Chimera strange behavior :))
I will send you my input files off-line.
Best wishes,
Marek
Dne Tue, 15 Jul 2014 20:42:39 +0200 Eric Pettersen <pett@cgl.ucsf.edu> napsal/-a:
Hi Marek, I'm glad my suggestion worked. I agree with you assessment, which is why I was hoping you could provide data files for testing. Nonetheless, I have gone ahead and made changes such that for formats where explicit topology files are provided, such as Amber, Chimera "trusts" the connectivity and does not change excessively long bonds into pseudobonds. Those changes will be in tonight's daily build and I'm hoping you can test them and let me know if they work. Look for daily builds dated July 15th or later.
--Eric
On Jul 15, 2014, at 4:08 AM, "Marek Maly" <marek.maly@ujep.cz> wrote:
Hi Eric, thank you for the prompt and useful answer.
The approach you suggested works !
But I already identified the source of the problem.
In my Amber MD trajectory, there was also the first(starting) configuration of my protein (that from the *.inpcrd file).
This starting configuration was not so precise as was obtained by the electron microscopy (not by X-ray or NMR technique).
From this reason distances of some of the sulfur atoms which should be connected by disulfide bond was not reasonable in this starting configuration but higher like 3 A or 6 A.
So in spite the fact that in the Amber topology (*.prmtop) file were clearly written all the disulfide bonds, Chimera evidently ignored those bonds in sulfur atom pairs where the distance was not in the reasonable range (close to 2A). Instead perhaps Chimera considered, that there are some missing parts or so and used "dashed line" instead "stick". So this was the first Chimera "mistake" which should not happen if she just simply accepted the full topology information from the "*.prmtop" file.
Another problem was that even if in all consequent frames of the trajectory all the relevant sulfur atoms were in the reasonable distance (2-2.1 A - due to the fact that during the Amber simulation they were bonded), Chimera doesn't care about because she evaluate the overall structure just using the first frame of the trajectory.
I would suggest here, that Chimera should not take her own decisions in such cases when molecular topology is fully defined in input files (e.g. Amber *.prmtop, mol2 etc.).
Especially in case of the Amber *.prmtop file it is clear that structure should be OK, (e.g. without the missing parts) because otherwise this topology file would never be created.
If Chimera find something strange in the structure (even if this structure was fully determined in the input file/s), she can always write the proper warnings, and that should be enough in such specific cases, I think.
Best wishes,
Marek
Dne Mon, 14 Jul 2014 19:33:51 +0200 Eric Pettersen <pett@cgl.ucsf.edu> napsal/-a:
On Jul 14, 2014, at 8:57 AM, "Marek Maly" <marek.maly@ujep.cz> wrote:
Hellow,
I just visualized my Amber MD trajectory with protein containing several disulfide bonds. These bonds as well as all the other bonds are written in the given Amber topology (*.prmtop) file, which is read in by Chimera. Moreover the distance of sulfur atoms connected by disulfide bond is in all cases quite reasonable ( 2-2.1 A). In spite these facts Chimera draws some of those bonds in "stick" and some of them in "dashed line" style, even if one sets globally "stick" or "ball & stick" representation. Please see the attached illustrations.
Why Chimera is doing such unwanted differences in graphical representations here ?
It is possible, to force Chimera to draw all of those bonds unanimously in "stick" representation ?
Hi Marek, My best guess is that Chimera is mistakenly converting some of the the disulphide bonds into "missing structure" pseudobonds, which are supposed to be used to represent parts of a chain that missing because is couldn't be located in the experimental data -- which obviously is not the case here! The workaround to make those pseudobonds be depicted as stick is to open the Pseudobond Panel (in Tools/General Controls) and the double click on the "missing structure" group to bring up its attribute inspector. In that inspector, click the check box next to "Component Pseudobond Attributes" to reveal those options and then change "bond style" from wire to stick. There is an equivalent command: "setattr pb drawMode 1". Obviously the better solution is to have Chimera not make this mistake in the first place. As far as I know it shouldn't be already. So, I would need your prmtop file and at least a subset of your coordinate file in order to investigate. Also, what version of Chimera are you using?
--Eric
Eric Pettersen UCSF Computer Graphics Lab http://www.cgl.ucsf.edu
__________ Informace od ESET NOD32 Antivirus, verze databaze 10094 (20140714) __________
Tuto zpravu proveril ESET NOD32 Antivirus.
-- Tato zpráva byla vytvořena převratným poštovním klientem Opery: http://www.opera.com/mail/
__________ Informace od ESET NOD32 Antivirus, verze databaze 10101 (20140715) __________
Tuto zpravu proveril ESET NOD32 Antivirus.
-- Tato zpráva byla vytvořena převratným poštovním klientem Opery: http://www.opera.com/mail/
OK, thanks again, Best wishes, Marek Dne Tue, 15 Jul 2014 23:38:10 +0200 Eric Pettersen <pett@cgl.ucsf.edu> napsal/-a:
Thanks for the input files. I'm happy to report the problem is indeed fixed.
--Eric
On Jul 15, 2014, at 1:57 PM, "Marek Maly" <marek.maly@ujep.cz> wrote:
Hi Eric, thanks for your decisions.
Sorry that I forget to send my files for testing. I was fully satisfied just with the complete explanation of this Chimera strange behavior :))
I will send you my input files off-line.
Best wishes,
Marek
Dne Tue, 15 Jul 2014 20:42:39 +0200 Eric Pettersen <pett@cgl.ucsf.edu> napsal/-a:
Hi Marek, I'm glad my suggestion worked. I agree with you assessment, which is why I was hoping you could provide data files for testing. Nonetheless, I have gone ahead and made changes such that for formats where explicit topology files are provided, such as Amber, Chimera "trusts" the connectivity and does not change excessively long bonds into pseudobonds. Those changes will be in tonight's daily build and I'm hoping you can test them and let me know if they work. Look for daily builds dated July 15th or later.
--Eric
On Jul 15, 2014, at 4:08 AM, "Marek Maly" <marek.maly@ujep.cz> wrote:
Hi Eric, thank you for the prompt and useful answer.
The approach you suggested works !
But I already identified the source of the problem.
In my Amber MD trajectory, there was also the first(starting) configuration of my protein (that from the *.inpcrd file).
This starting configuration was not so precise as was obtained by the electron microscopy (not by X-ray or NMR technique).
From this reason distances of some of the sulfur atoms which should be connected by disulfide bond was not reasonable in this starting configuration but higher like 3 A or 6 A.
So in spite the fact that in the Amber topology (*.prmtop) file were clearly written all the disulfide bonds, Chimera evidently ignored those bonds in sulfur atom pairs where the distance was not in the reasonable range (close to 2A). Instead perhaps Chimera considered, that there are some missing parts or so and used "dashed line" instead "stick". So this was the first Chimera "mistake" which should not happen if she just simply accepted the full topology information from the "*.prmtop" file.
Another problem was that even if in all consequent frames of the trajectory all the relevant sulfur atoms were in the reasonable distance (2-2.1 A - due to the fact that during the Amber simulation they were bonded), Chimera doesn't care about because she evaluate the overall structure just using the first frame of the trajectory.
I would suggest here, that Chimera should not take her own decisions in such cases when molecular topology is fully defined in input files (e.g. Amber *.prmtop, mol2 etc.).
Especially in case of the Amber *.prmtop file it is clear that structure should be OK, (e.g. without the missing parts) because otherwise this topology file would never be created.
If Chimera find something strange in the structure (even if this structure was fully determined in the input file/s), she can always write the proper warnings, and that should be enough in such specific cases, I think.
Best wishes,
Marek
Dne Mon, 14 Jul 2014 19:33:51 +0200 Eric Pettersen <pett@cgl.ucsf.edu> napsal/-a:
On Jul 14, 2014, at 8:57 AM, "Marek Maly" <marek.maly@ujep.cz> wrote:
Hellow,
I just visualized my Amber MD trajectory with protein containing several disulfide bonds. These bonds as well as all the other bonds are written in the given Amber topology (*.prmtop) file, which is read in by Chimera. Moreover the distance of sulfur atoms connected by disulfide bond is in all cases quite reasonable ( 2-2.1 A). In spite these facts Chimera draws some of those bonds in "stick" and some of them in "dashed line" style, even if one sets globally "stick" or "ball & stick" representation. Please see the attached illustrations.
Why Chimera is doing such unwanted differences in graphical representations here ?
It is possible, to force Chimera to draw all of those bonds unanimously in "stick" representation ?
Hi Marek, My best guess is that Chimera is mistakenly converting some of the the disulphide bonds into "missing structure" pseudobonds, which are supposed to be used to represent parts of a chain that missing because is couldn't be located in the experimental data -- which obviously is not the case here! The workaround to make those pseudobonds be depicted as stick is to open the Pseudobond Panel (in Tools/General Controls) and the double click on the "missing structure" group to bring up its attribute inspector. In that inspector, click the check box next to "Component Pseudobond Attributes" to reveal those options and then change "bond style" from wire to stick. There is an equivalent command: "setattr pb drawMode 1". Obviously the better solution is to have Chimera not make this mistake in the first place. As far as I know it shouldn't be already. So, I would need your prmtop file and at least a subset of your coordinate file in order to investigate. Also, what version of Chimera are you using?
--Eric
Eric Pettersen UCSF Computer Graphics Lab http://www.cgl.ucsf.edu
__________ Informace od ESET NOD32 Antivirus, verze databaze 10094 (20140714) __________
Tuto zpravu proveril ESET NOD32 Antivirus.
-- Tato zpráva byla vytvořena převratným poštovním klientem Opery: http://www.opera.com/mail/
__________ Informace od ESET NOD32 Antivirus, verze databaze 10101 (20140715) __________
Tuto zpravu proveril ESET NOD32 Antivirus.
-- Tato zpráva byla vytvořena převratným poštovním klientem Opery: http://www.opera.com/mail/
__________ Informace od ESET NOD32 Antivirus, verze databaze 10101 (20140715) __________
Tuto zpravu proveril ESET NOD32 Antivirus.
-- Tato zpráva byla vytvořena převratným poštovním klientem Opery: http://www.opera.com/mail/
participants (2)
-
 Eric Pettersen
Eric Pettersen -
 Marek Maly
Marek Maly