
Hello, After using autodock and obtaining the HBonds, it seems that if I run the simulation with the exact same parameters for receptor search volume, the HBonds numbers change along with some other information. I have added the file images to show. Is this supposed to be happening or is this a user error? First run: Second Run with same parameters: Thank you! -- Dustin Park University of Illinois Urbana-Champaign (M. Eng - Bioengineering) Mobile: *224.623.3626*
{kind=link}
{kind=link}
{kind=link}
Hello, Sorry, I had inserted the wrong second image. I attached another image. On Wed, Apr 11, 2018 at 3:32 PM, Dustin Park <dpark61@illinois.edu> wrote:
Hello,
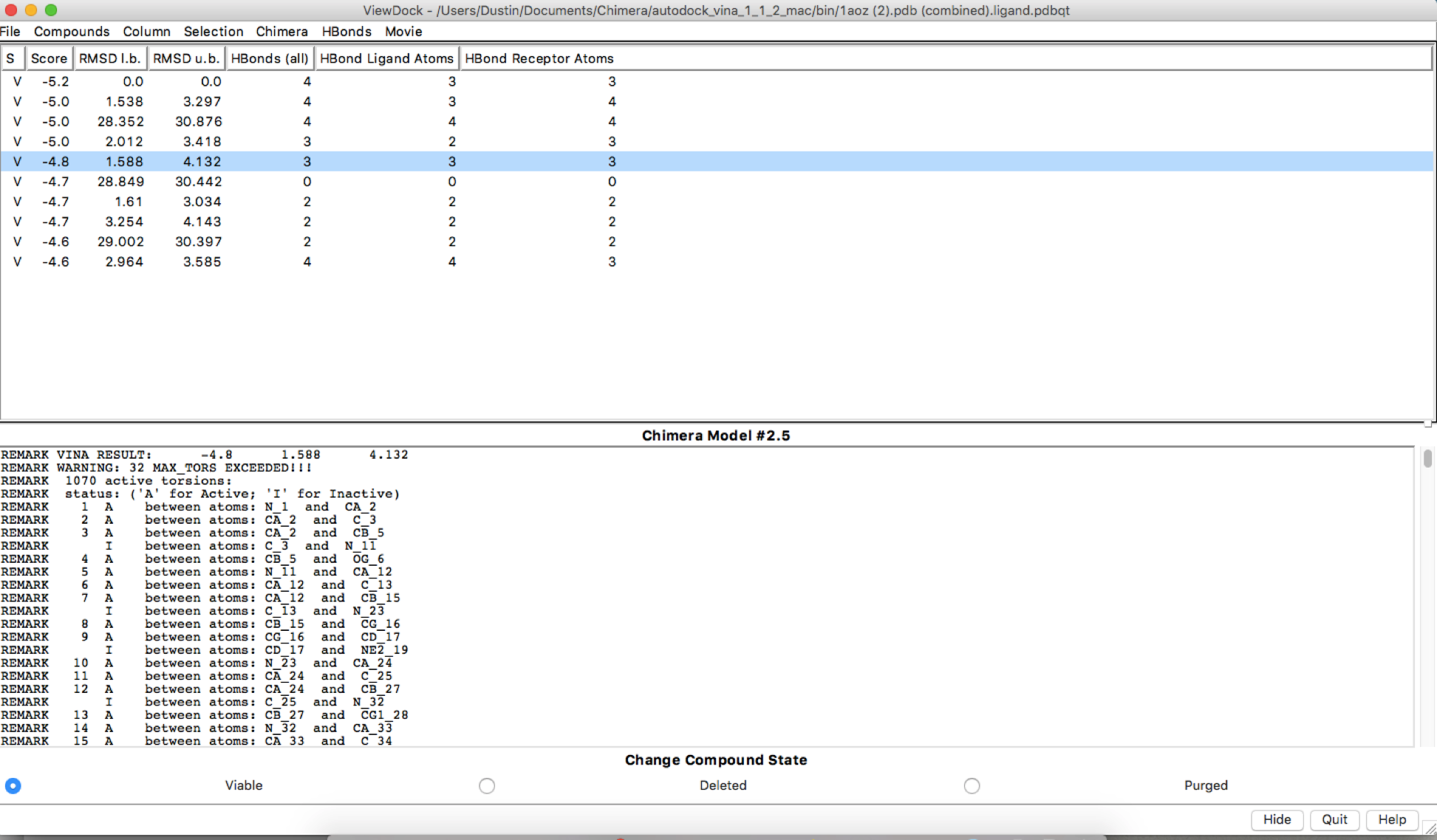
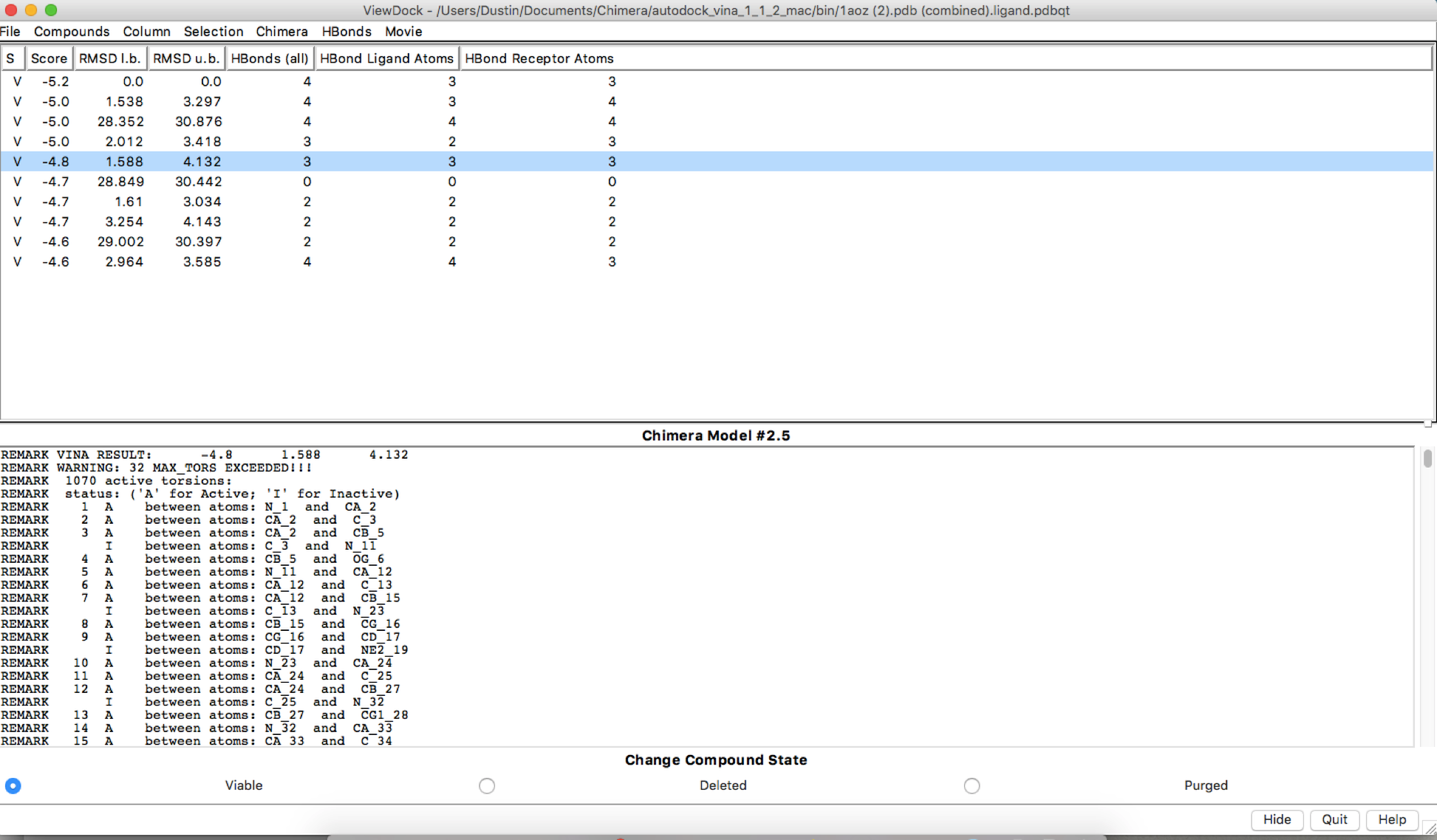
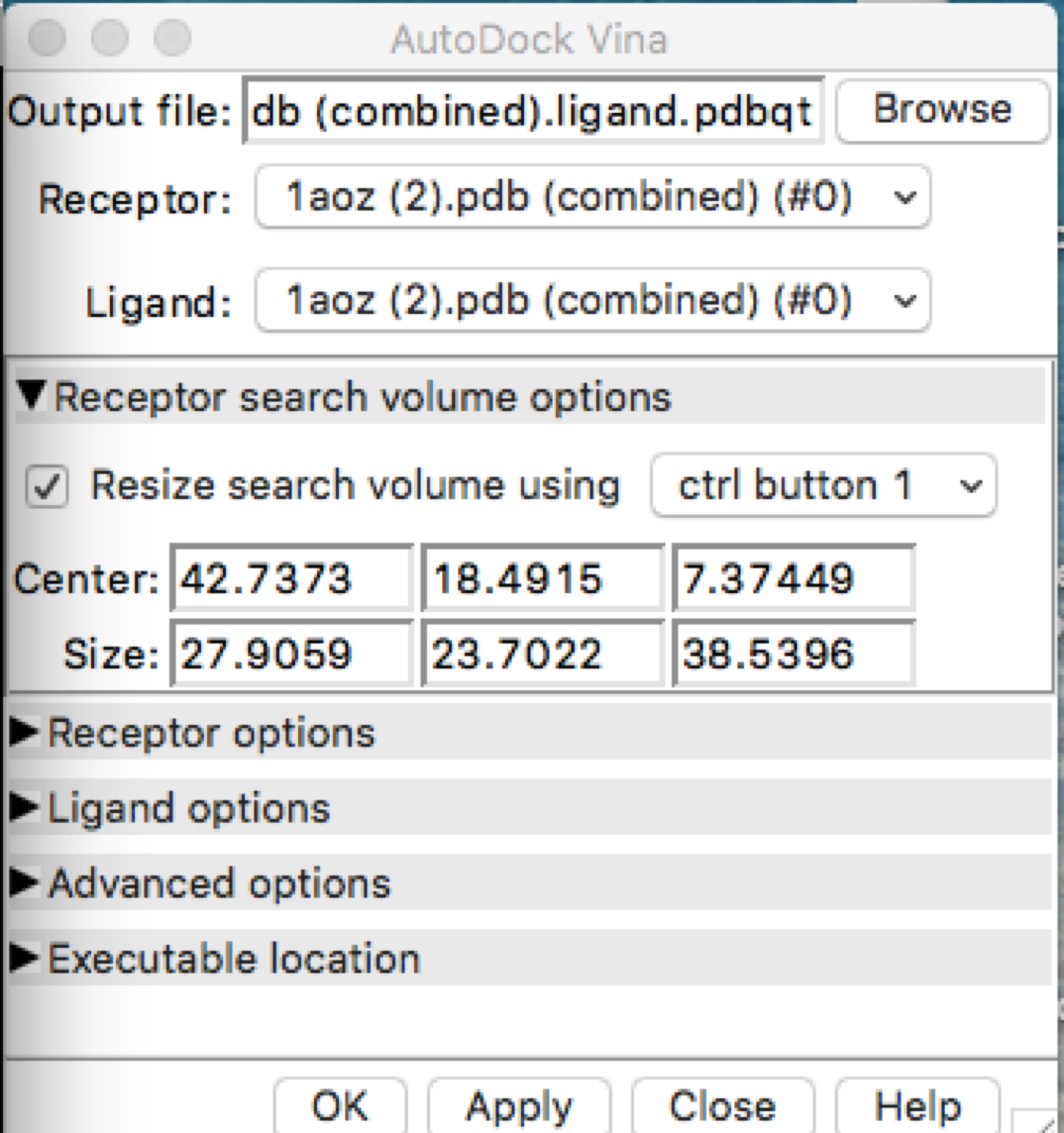
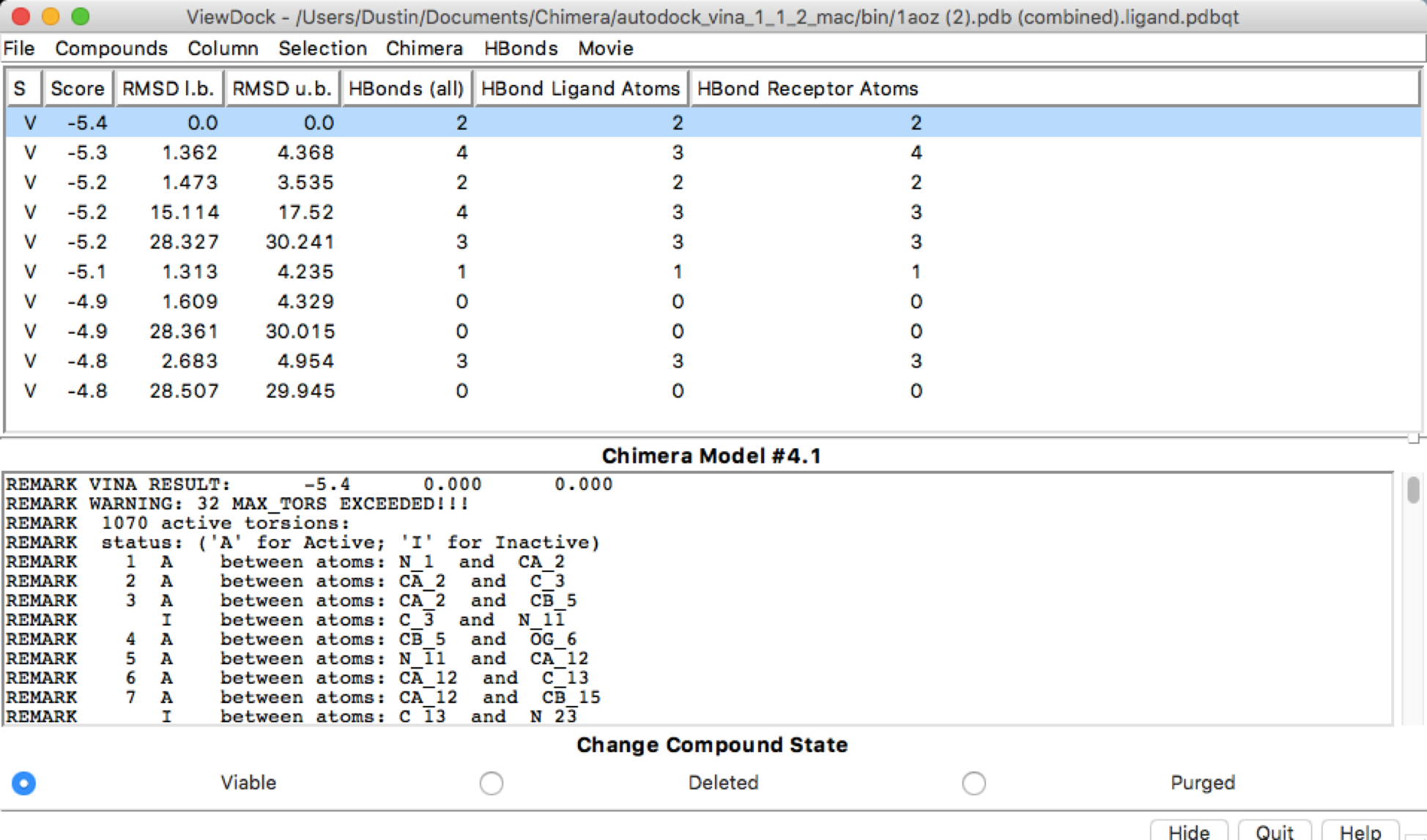
After using autodock and obtaining the HBonds, it seems that if I run the simulation with the exact same parameters for receptor search volume, the HBonds numbers change along with some other information. I have added the file images to show. Is this supposed to be happening or is this a user error?
First run:
Second Run with same parameters:
Thank you!
-- Dustin Park University of Illinois Urbana-Champaign (M. Eng - Bioengineering) Mobile: *224.623.3626*
-- Dustin Park University of Illinois Urbana-Champaign (M. Eng - Bioengineering) Mobile: *224.623.3626*
{kind=link}
{kind=link}
{kind=link}
{kind=link}

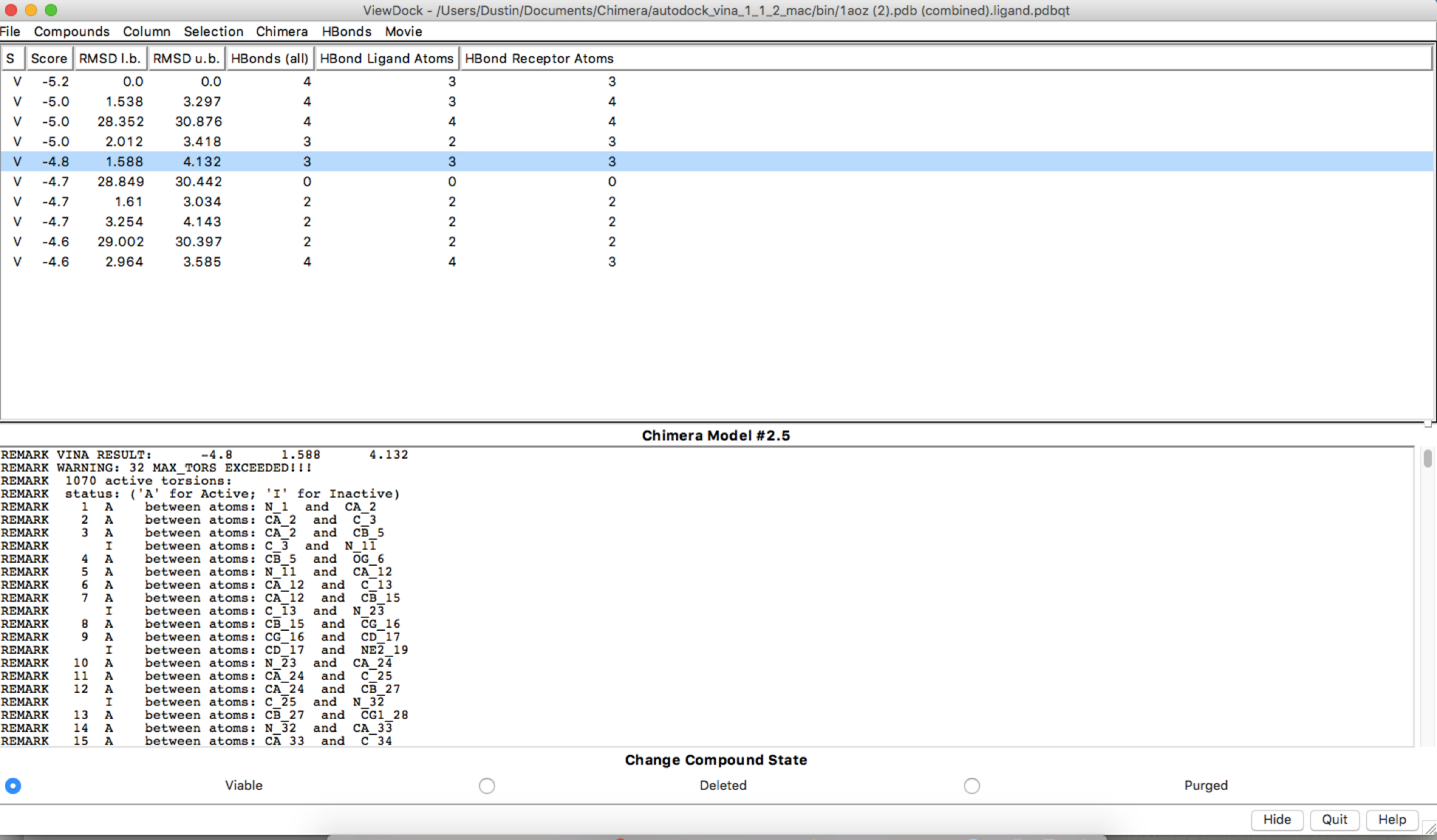
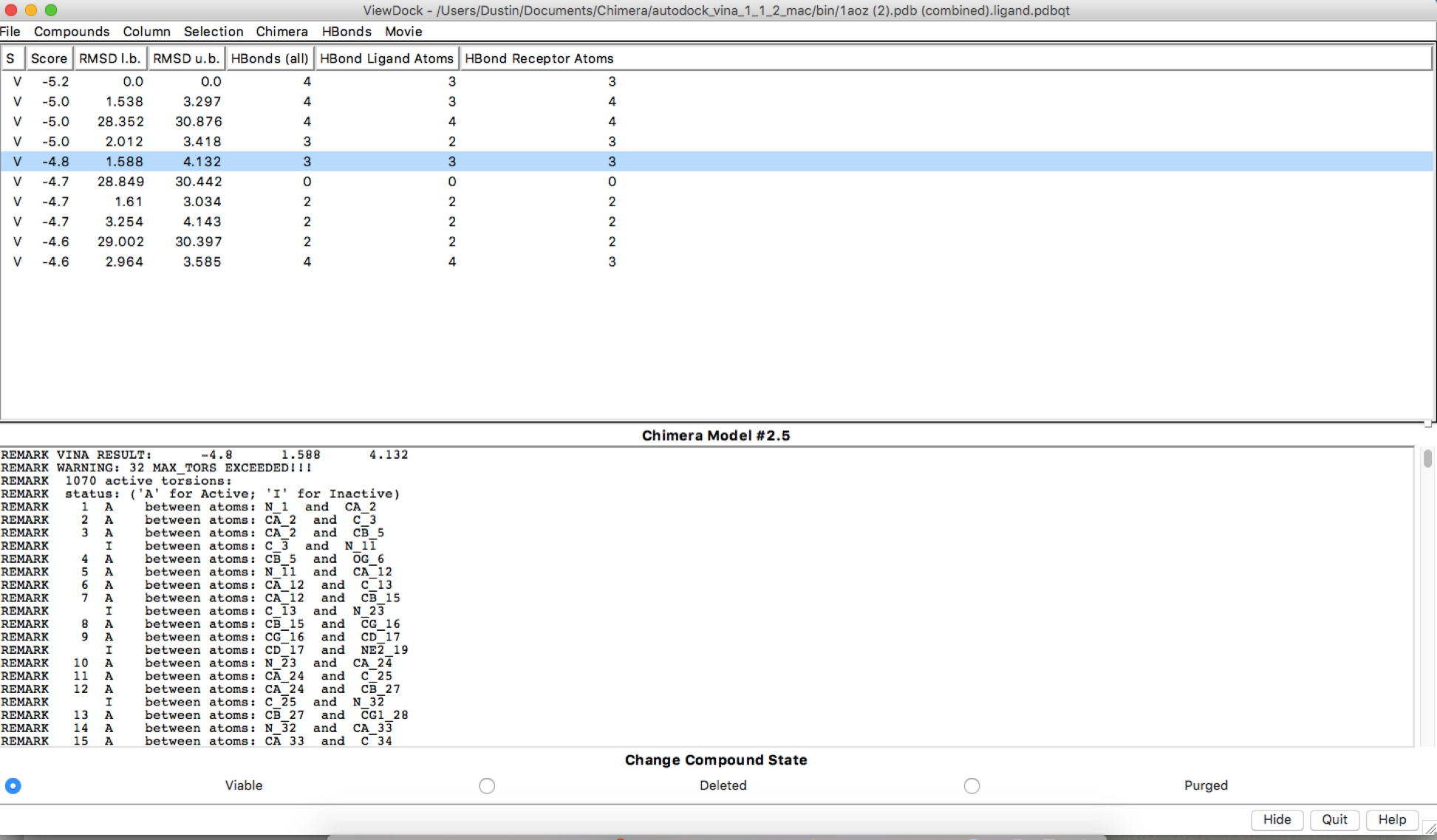
Hi Dustin, There may be some stochasticity in the Vina sampling (e.g. use of random number generator), but I don’t know for certain. We are not the Vina developers, you may want to delve into their documention and publications. AutoDock Vina website: <http://vina.scripps.edu/> Of course, if the output positions are slightly different, it makes sense that the numbers of H-bonds are different. HOWEVER… your dialog shows that the ligand and the receptor are the same model. This makes no sense. I assume you aren’t really trying to dock a protein to itself. There are other indicators of this crazy situation, like the results saying that the ligand has 1070 active torsions (rotatable bonds). As I understand it, Vina is mainly for docking small molecules to proteins. Even if you wanted to try to use it for protein-protein docking, the amount of sampling available through this web service is quite small and unsuited to the task. If you really did want to do protein-protein docking, you may want to look into other available tools. Best, Elaine ----- Elaine C. Meng, Ph.D. UCSF Chimera(X) team Department of Pharmaceutical Chemistry University of California, San Francisco
On Apr 11, 2018, at 1:54 PM, Dustin Park <dpark61@illinois.edu> wrote:
Hello,
Sorry, I had inserted the wrong second image. I attached another image.
<image.png>
On Wed, Apr 11, 2018 at 3:32 PM, Dustin Park <dpark61@illinois.edu> wrote: Hello,
After using autodock and obtaining the HBonds, it seems that if I run the simulation with the exact same parameters for receptor search volume, the HBonds numbers change along with some other information. I have added the file images to show. Is this supposed to be happening or is this a user error?
<image.png> First run: <image.png> Second Run with same parameters: <image.png>
Thank you!
participants (2)
-
 Dustin Park
Dustin Park -
 Elaine Meng
Elaine Meng