Re: [Chimera-users] Chimera Surface Calculation

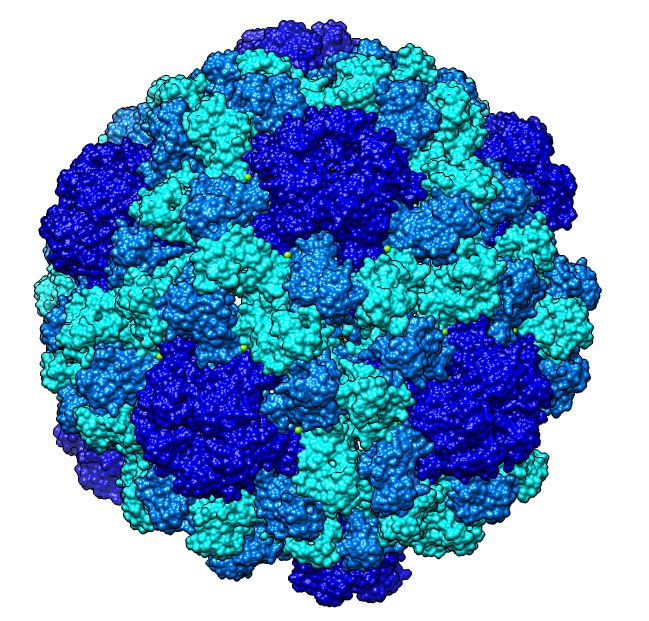
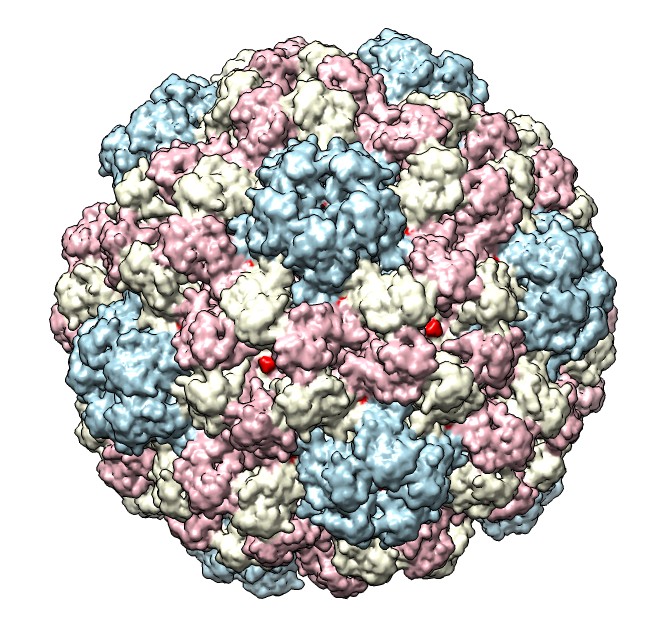
Hi Robert, Unfortunately the MSMS molecular surface calculation code has bugs and so it fails to compute surfaces for most large (>10,000 atom) PDB models. The surface calculation has essentially no chance of working with a model of 110,000 atoms such as your half-virus. That said I used the 64-bit Mac Chimera and split the model by chains and it surfaced fine -- see attached image. The Windows Chimera versions have the most surface calculation failures especially 64-bit Windows Chimera, and Mac and Linux are substantially better. So I made the image bmv.jpg with "split #0; surf; rainbow model". After the split, it is odd that many of the subunits have no atoms shown. I used Actions / Atoms / Show to show them. If you don't require a solvent excluded surface (rolling a ball over the atoms to get the surface), you can use a Gaussian surface. The second image bmv5.jpg was made without splitting the model using a 5 Angstrom density map. molmap #0 5 model #1 volume #1 step 1 color lightblue :.A color lightyellow :.B color pink :.C color red :.D-K scolor #1 zone #0 range 10 Tom
Hi Tom, I am a post doc at Indiana University in the Biochemistry department, and I was at the chimera tutorial you gave to our department through David Morgan. I wasn't sure who to write with this question, so I thought I would start with you but if this is more appropriate for someone else, just let me know.
Forever ago I used Viper to create a 60 subunit (or 1/2) capsid for BMV, and made several models for publications. That .pdb file that I made no longer works within Chimera, and I have literally tried everything to get it to work. Additionally, it seems Viper no longer lets you make those .pdb files anymore. I have tried changing the atomic radii probe radius, the vertex density, set the disjoint surfaces to false, and every combination of those things. Additionally, when I use the split command, it either only shows a weird surface or no surface at all, and almost completely obliterates the original structure. If you know another way to do this, I would really appreciate any advice here as I have spent hours reading through forums. I attached the .pdb to this email. Again I really appreciate your help.
Thanks in advance, Robert
{kind=link}
{kind=link}

On Tue, 17 Jul 2012 10:34:54 -0700 Tom Goddard <goddard@sonic.net> wrote:
Hi Robert,
Unfortunately the MSMS molecular surface calculation code has bugs and so it fails to compute surfaces for most large (>10,000 atom) PDB models. The surface calculation has essentially no chance of working with a model of 110,000 atoms such as your half-virus. That said I used the 64-bit Mac Chimera and split the model by chains and it surfaced fine -- see attached image. The Windows Chimera versions have the most surface calculation failures especially 64-bit Windows Chimera, and Mac and Linux are substantially better.
So I made the image bmv.jpg with "split #0; surf; rainbow model". ...
I swear this looks like a piece of broccoli seen from above (and through blue--or ??--colored glasses ;-) ... Kenward -- In a completely rational society, the best of us would aspire to be _teachers_ and the rest of us would have to settle for something less, because passing civilization along from one generation to the next ought to be the highest honor and the highest responsibility anyone could have. - Lee Iacocca

Hi All, I am a new user of Chimera and need to quickly prepare an image in which the hypervariable regions of IgH and IgH are colored differently from the constant regions. For example I want to color these ranges in the two IgG chains: H chain: blue: 25-30, 51-59, 97-101 L chain: green: 29-34, 50-57, 93 - 95C I would greatly appreciate some help in learning how to do this. Thanks, Peter Hornbeck

Hi Peter, There are several examples of command-line specification of models, chains, residues, atoms at the end the Quick Reference (2-page PDF): <http://www.cgl.ucsf.edu/chimera/current/docs/UsersGuide/quickref.pdf> ...and the full explanation is in the "atom specification" page of the manual: <http://www.cgl.ucsf.edu/chimera/current/docs/UsersGuide/midas/frameatom_spec...> For example, you could use commands like: alias group2 #0:29-34.L,50-57.L,90-95C.L color lime green,r group2 Actually it could be done in one command without aliasing, but I personally like to do the aliasing because it makes it easier to try different colors without typing all the residue numbers again. The ",r" after the color name is an example of using the command to color ribbons only (not atoms/bonds, surface patches, etc.). If you omit that, it will color all the representations of those residues. See the "color" manual page: <http://www.cgl.ucsf.edu/chimera/current/docs/UsersGuide/midas/color.html> Commands are really the best for this kind of thing. However, it could also be done via GUIs only. You could use Sequence (under Favorites) to select stretches of residues in the sequence view with the mouse, then use the Color Actions dialog, menu: Actions... Color... all options...). I hope this helps, Elaine ----- Elaine C. Meng, Ph.D. UCSF Computer Graphics Lab (Chimera team) and Babbitt Lab Department of Pharmaceutical Chemistry University of California, San Francisco On Jul 18, 2012, at 5:51 AM, Peter Hornbeck wrote:
Hi All, I am a new user of Chimera and need to quickly prepare an image in which the hypervariable regions of IgH and IgH are colored differently from the constant regions.
For example I want to color these ranges in the two IgG chains:
H chain: blue: 25-30, 51-59, 97-101 L chain: green: 29-34, 50-57, 93 - 95C
I would greatly appreciate some help in learning how to do this. Thanks, Peter Hornbeck list Chimera-users@cgl.ucsf.edu http://plato.cgl.ucsf.edu/mailman/listinfo/chimera-users
participants (4)
-
 Elaine Meng
Elaine Meng -
 Kenward Vaughan
Kenward Vaughan -
 Peter Hornbeck
Peter Hornbeck -
 Tom Goddard
Tom Goddard