Distance tool: Line thickness of the number (of angstroms) in the picture

Hi, The number that shows up after creating a distance is very thin and can hardly be seen. I couldn't find how to make it thicker (I did change the actual line thickness in the menu). I also tried in the preference menu that you pointed to a user a while ago but it didn't work for me. How can I change it? This is on the Windows 64b nightly build from 17 October. Thanks. Boaz Boaz Shaanan, Ph.D. Dept. of Life Sciences Ben-Gurion University of the Negev Beer-Sheva 84105 Israel E-mail: bshaanan@bgu.ac.il Phone: 972-8-647-2220 Skype: boaz.shaanan Fax: 972-8-647-2992 or 972-8-646-1710 ________________________________________ From: Chimera-users [chimera-users-bounces@cgl.ucsf.edu] on behalf of chimera-users-request@cgl.ucsf.edu [chimera-users-request@cgl.ucsf.edu] Sent: Saturday, October 21, 2017 1:17 AM To: chimera-users@cgl.ucsf.edu Subject: Chimera-users Digest, Vol 174, Issue 28 Send Chimera-users mailing list submissions to chimera-users@cgl.ucsf.edu To subscribe or unsubscribe via the World Wide Web, visit http://plato.cgl.ucsf.edu/mailman/listinfo/chimera-users or, via email, send a message with subject or body 'help' to chimera-users-request@cgl.ucsf.edu You can reach the person managing the list at chimera-users-owner@cgl.ucsf.edu When replying, please edit your Subject line so it is more specific than "Re: Contents of Chimera-users digest..." Today's Topics: 1. Re: Trouble with Volume surface and active model not aligned (Elaine Meng) 2. Re: Selecting residues in chains defined by segname (Francesco Pietra) 3. Re: Selecting residues in chains defined by segname (Elaine Meng) 4. Re: Trouble with Volume surface and active model not aligned (Phan Trieu Truong) ---------------------------------------------------------------------- Message: 1 Date: Fri, 20 Oct 2017 13:41:34 -0700 From: Elaine Meng <meng@cgl.ucsf.edu> To: Phan Trieu Truong <truongp@uga.edu> Cc: "chimera-users@cgl.ucsf.edu" <chimera-users@cgl.ucsf.edu> Subject: Re: [Chimera-users] Trouble with Volume surface and active model not aligned Message-ID: <5596D972-35F9-4643-8CDB-6F7D4DE590FD@cgl.ucsf.edu> Content-Type: text/plain; charset=utf-8 Hi Phan, I?m not sure what happened either, since your images show the ?A? (active for motion) boxes in the Model Panel are checked for both the molecule and the orbitals, meaning that using the mouse should move both of them together. You could try command: reset ? to put them back together. If that doesn?t work, you may have to start over by quitting and reopening the files. However, if you didn?t uncheck the ?A? boxes or the ?Active models? boxes below the Command Line, mouse movements should always move them both together. Maybe you accidentally had one of the A(ctive) boxes unchecked and moved the other model. In that case, reset will work. I hope this helps, Elaine ----- Elaine C. Meng, Ph.D. UCSF Chimera(X) team Department of Pharmaceutical Chemistry University of California, San Francisco
On Oct 20, 2017, at 8:57 AM, Phan Trieu Truong <truongp@uga.edu> wrote:
Hi,
I have been using Chimera to visualize MO orbital on molecules from ORCA calculations. I have been able to visualize the MO orbitals as a surface volume from cube files. However, I just found out that when I try to reorient the molecule with the surface volume, they are no longer aligned. I don't know what I did to cause this. Any help is much appreciated.
Attached is the picture of the molecules before and after I tried to reorient the molecule with the surface volume.
Thank you, Phan <Before and after reorientation.jpg.png>_______________________________________________ Chimera-users mailing list: Chimera-users@cgl.ucsf.edu Manage subscription: http://plato.cgl.ucsf.edu/mailman/listinfo/chimera-users
------------------------------ Message: 2 Date: Fri, 20 Oct 2017 23:37:32 +0200 From: Francesco Pietra <chiendarret@gmail.com> To: Eric Pettersen <pett@cgl.ucsf.edu> Cc: chimera <chimera-users@cgl.ucsf.edu> Subject: Re: [Chimera-users] Selecting residues in chains defined by segname Message-ID: <CAEv0nmsDA8dd+hQ8H_t9uK8-LntPucKQ7UiiqVybva3XWdBgjA@mail.gmail.com> Content-Type: text/plain; charset="utf-8" A pity, in my view. As more and more complex proteins are being examined (thanks to faster clusters), working without segname would be practically impossible in the frame of today pdb files. For example, how visualizing the trajectory of a particular ligand in a 24-chain protein assembly? I can do that with vmd, but at the price of a less clear-cut trajectory and poorer graphics. But I understand that there may be different viewpoints. Cheers francesco On Fri, Oct 20, 2017 at 8:36 PM, Eric Pettersen <pett@cgl.ucsf.edu> wrote:
Realistically no. Too many other priorities. The gap will eventually get filled in in ChimeraX, but even that will be awhile.
?Eric
On Oct 19, 2017, at 1:24 PM, Francesco Pietra <chiendarret@gmail.com> wrote:
Hi Eric:
Any plan to fill this gap?
thanks
francesco
On Thu, Oct 19, 2017 at 7:59 PM, Eric Pettersen <pett@cgl.ucsf.edu> wrote:
Hi Francseco, Unfortunately, segment IDs are not preserved by the trajectory reader.
?Eric
Eric Pettersen UCSF Computer Graphics Lab
On Oct 19, 2017, at 9:53 AM, Francesco Pietra <chiendarret@gmail.com> wrote:
Hi Elaine: While, as I wrote, the commands for segname did work fine with .psf/.pdb namd files, in contrast, with .psf/.dcd files (movie)
select @/pdbSegment=C1
selects all chains of the protein assembly, including ligands. The same occurs with
select @/pdbSegment=O2C1
where O2C1 is the segname of the molecule dioxygen associated with chain.
This occurs both on my desktop and on a large-memory nextscale cluster on remote visualization (the latter is the actual interest)
Do you know of any remedy?
thanks a lot
francesco ---------- Forwarded message ---------- From: Francesco Pietra <chiendarret@gmail.com> Date: Mon, Oct 16, 2017 at 7:12 PM Subject: Re: [Chimera-users] Selecting residues in chains defined by segname To: UCSF Chimera Mailing List <chimera-users@cgl.ucsf.edu>
Hi Elaine:
Great!
thank you francesco
On Mon, Oct 16, 2017 at 5:58 PM, Elaine Meng <meng@cgl.ucsf.edu> wrote:
Hi Francesco, The symbol for intersection is ?&? ... in other words, you could use
select :17 & @/pdbSegment=A1
Intersection and union symbols are explained here: <http://www.rbvi.ucsf.edu/home/meng/docs/UsersGuide/midas/at om_spec.html#combinations>
I hope this helps, Elaine
On Oct 16, 2017, at 12:29 AM, Francesco Pietra <chiendarret@gmail.com> wrote:
Hi Elaine: That works fine. However, I was unable to extend your suggestions to pick up a specific residue within a specific chain. Neither "select :17 @/pdbSegment=A1" nor "select @/pdbSegment=A1 :17" are valid commands (obviously expected).
On the other hand, with such complex situations, it is Xplor, with its segname features, that helps.
Should you need a pdb fine with segname, I could attach a simple one, with a single chain.
On Sun, Oct 15, 2017 at 8:04 PM, Elaine Meng <meng@cgl.ucsf.edu> wrote: Hi Francesco, Although I don?t have an example file with segnames to try myself, I?m told you can specify by the atom attribute pdbSegment, e.g.
select @/pdbSegment=A1 color red @/pdbSegment=F3
I hope this helps, Elaine
On Oct 15, 2017, at 10:40 AM, Francesco Pietra < chiendarret@gmail.com> wrote:
Hi Elaine:
I am referring to Oct 28, 2005, at 9:50 AM, Eric Gillitzer wrote: and your answer:
command: select :45.a-d > or > command: select :45.* > > Or, to select residue 45 in just chains A and D: > > command: select :45.a,45.d
I have a more complex case, where chains are defined by segname, for example
A1 A2 A3 A4 A5 etc
while the standard PDB definition is "A" for all them.
The same for standard "B", "C" etc.
As I want to display a movie of ligand pathways, where the ligand moves from, say, "A1" to, say, "F3", I want in the first instance become able to select particular residues in particular chains, as defined by their segname.
Could you imagine a simple way not requiring selection by atom numbers? Thanks francesco pietra
_______________________________________________ Chimera-users mailing list: Chimera-users@cgl.ucsf.edu Manage subscription: http://plato.cgl.ucsf.edu/mail man/listinfo/chimera-users
_______________________________________________ Chimera-users mailing list: Chimera-users@cgl.ucsf.edu Manage subscription: http://plato.cgl.ucsf.edu/mail man/listinfo/chimera-users

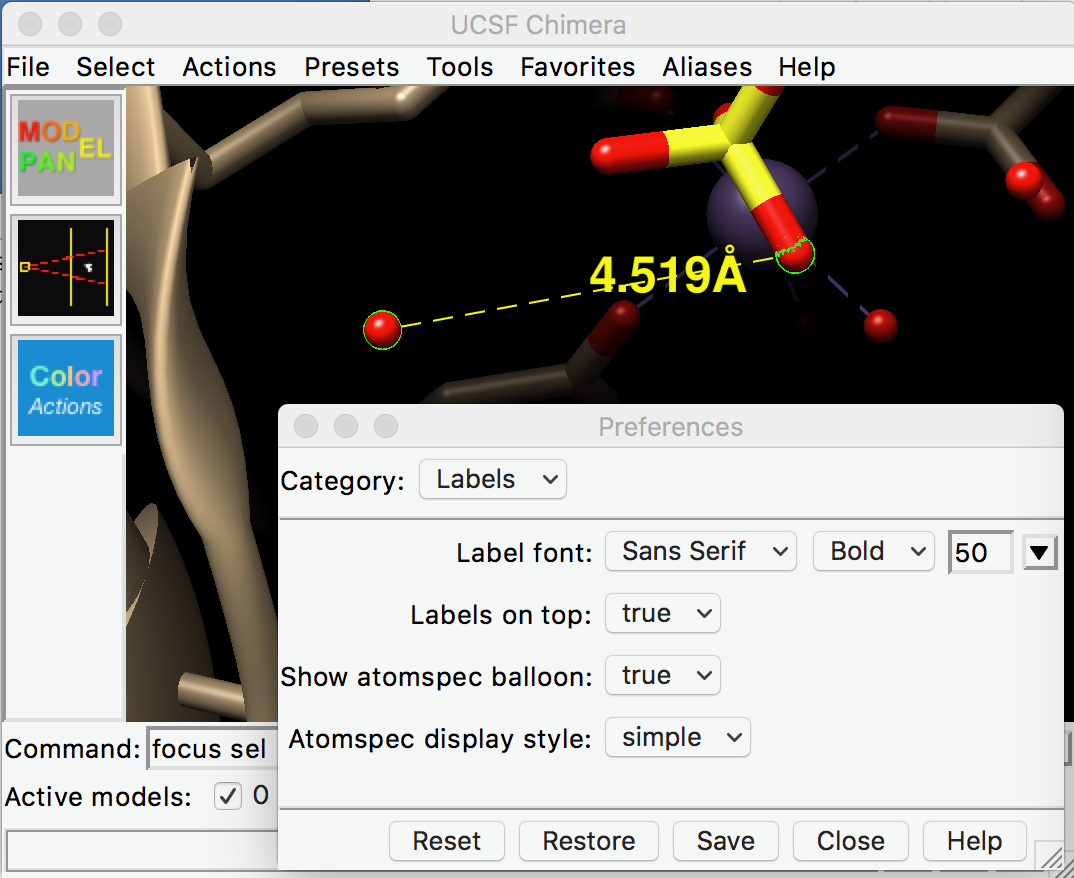
Hi Boaz, The only user interface to the label font size and style is the Preferences (open from Favorites menu), category: Labels. You could change the size number and make the style bold. This is for all “3D” atom and distance labels that move along with the structure; size/font of 2D labels that don’t move with the structure can be set individually. Looks like the Labels Preferences size menu only goes up to 36, but you can enter some bigger number… for example, I entered 50 (see screenshot attached). Smaller size changes were barely perceptible on my high-resolution screen; maybe that is what happened for you. If that doesn’t work, I’m at a loss… maybe some weird rendering problem? I hope this helps, Elaine ----- Elaine C. Meng, Ph.D. UCSF Chimera(X) team Department of Pharmaceutical Chemistry University of California, San Francisco
On Oct 21, 2017, at 4:31 AM, Boaz Shaanan <bshaanan@bgu.ac.il> wrote:
Hi, The number that shows up after creating a distance is very thin and can hardly be seen. I couldn't find how to make it thicker (I did change the actual line thickness in the menu). I also tried in the preference menu that you pointed to a user a while ago but it didn't work for me. How can I change it? This is on the Windows 64b nightly build from 17 October. Thanks. Boaz
{kind=link}
participants (2)
-
 Boaz Shaanan
Boaz Shaanan -
 Elaine Meng
Elaine Meng