
Dear Chimera users, I have a pdb file of my protein, but not showing the binding ligand (Heme). I know where the binding motif is and I am wondering is there a way I can add ligand to the binding motif and write out the new pdb file in Chimera? Best, Yangqi -- Yangqi Gu Graduate Student Malvankar Lab Yale University, West Campus

Hi Yangqi, Of course you can open multiple structures and move them how you like and and then save again as PDB or session. See this previous post for “manual placement of ligand” <http://plato.cgl.ucsf.edu/pipermail/chimera-users/2011-June/006473.html> Another way is to open your protein pdb file, then open a similar pdb that does have the heme in it, superimpose the two models, and then delete the other protein atoms so that only the heme is left together with your protein. You could just save the PDB file with both models in it, or you could combine them into one model first. How to superimpose structures: <http://www.rbvi.ucsf.edu/chimera/docs/UsersGuide/superposition.html> Use menu: Help… Search Documentation to search for “delete” “combine” “save PDB” etc. I hope this helps, Elaine ----- Elaine C. Meng, Ph.D. UCSF Chimera(X) team Department of Pharmaceutical Chemistry University of California, San Francisco
On Dec 26, 2018, at 12:05 PM, Yangqi Gu <yangqi.gu@yale.edu> wrote:
Dear Chimera users, I have a pdb file of my protein, but not showing the binding ligand (Heme). I know where the binding motif is and I am wondering is there a way I can add ligand to the binding motif and write out the new pdb file in Chimera? Best, Yangqi

Dear Elaine How can I calculate the affinity and activity. Best Rgards On Wed, Dec 26, 2018 at 11:43 PM Elaine Meng <meng@cgl.ucsf.edu> wrote:
Hi Yangqi, Of course you can open multiple structures and move them how you like and and then save again as PDB or session.
See this previous post for “manual placement of ligand” <http://plato.cgl.ucsf.edu/pipermail/chimera-users/2011-June/006473.html>
Another way is to open your protein pdb file, then open a similar pdb that does have the heme in it, superimpose the two models, and then delete the other protein atoms so that only the heme is left together with your protein. You could just save the PDB file with both models in it, or you could combine them into one model first.
How to superimpose structures: <http://www.rbvi.ucsf.edu/chimera/docs/UsersGuide/superposition.html>
Use menu: Help… Search Documentation to search for “delete” “combine” “save PDB” etc.
I hope this helps, Elaine ----- Elaine C. Meng, Ph.D. UCSF Chimera(X) team Department of Pharmaceutical Chemistry University of California, San Francisco
On Dec 26, 2018, at 12:05 PM, Yangqi Gu <yangqi.gu@yale.edu> wrote:
Dear Chimera users, I have a pdb file of my protein, but not showing the binding ligand (Heme). I know where the binding motif is and I am wondering is there a way I can add ligand to the binding motif and write out the new pdb file in Chimera? Best, Yangqi
_______________________________________________ Chimera-users mailing list: Chimera-users@cgl.ucsf.edu Manage subscription: http://plato.cgl.ucsf.edu/mailman/listinfo/chimera-users
-- Assistant Prof.Dr. Adil Muala Dhumad University of Basrah, College of Education for Pure Sciences, Department of Chemistry
Dear Dr. Adil Muala Dhumad Ezirej, Sorry, Chimera does not calculate affinity or activity. Best, Elaine ----- Elaine C. Meng, Ph.D. UCSF Chimera(X) team Department of Pharmaceutical Chemistry University of California, San Francisco
On Dec 26, 2018, at 2:51 PM, dr. Adil Muala Dhumad Ezirej <dr.adilmualadhumad@gmail.com> wrote:
Dear Elaine How can I calculate the affinity and activity. Best Rgards
Dear Elaine Meng, Merry Christmas and Happy New Year. When the protein is added to the ligand (as in the attach picture) , the distance between them is remote. How can I get closer to ligand of the future protein. best regards Adil M. Dhumad On Thu, 27 Dec 2018 at 20:13, Elaine Meng <meng@cgl.ucsf.edu> wrote:
Dear Dr. Adil Muala Dhumad Ezirej,
Sorry, Chimera does not calculate affinity or activity.
Best, Elaine ----- Elaine C. Meng, Ph.D. UCSF Chimera(X) team Department of Pharmaceutical Chemistry University of California, San Francisco
On Dec 26, 2018, at 2:51 PM, dr. Adil Muala Dhumad Ezirej < dr.adilmualadhumad@gmail.com> wrote:
Dear Elaine How can I calculate the affinity and activity. Best Rgards
-- Assistant Prof.Dr. Adil Muala Dhumad University of Basrah, College of Education for Pure Sciences, Department of Chemistry
{kind=link}
Dear Adil M. Dhumad, Please see this previous post and links therein for suggestions on how to position the ligand: <http://plato.cgl.ucsf.edu/pipermail/chimera-users/2018-December/015306.html> Best regards, Elaine ----- Elaine C. Meng, Ph.D. UCSF Chimera(X) team Department of Pharmaceutical Chemistry University of California, San Francisco
On Dec 30, 2018, at 1:51 PM, dr. Adil Muala Dhumad Ezirej <dr.adilmualadhumad@gmail.com> wrote:
Dear Elaine Meng, Merry Christmas and Happy New Year. When the protein is added to the ligand (as in the attach picture) , the distance between them is remote. How can I get closer to ligand of the future protein. best regards
Adil M. Dhumad
Dear Elaine Meng, please help me to solution this problem. I'm dont know what's the problem in attach case. Best regards On Mon, 31 Dec 2018 at 20:43, Elaine Meng <meng@cgl.ucsf.edu> wrote:
Dear Adil M. Dhumad, Please see this previous post and links therein for suggestions on how to position the ligand:
< http://plato.cgl.ucsf.edu/pipermail/chimera-users/2018-December/015306.html
Best regards, Elaine ----- Elaine C. Meng, Ph.D. UCSF Chimera(X) team Department of Pharmaceutical Chemistry University of California, San Francisco
On Dec 30, 2018, at 1:51 PM, dr. Adil Muala Dhumad Ezirej < dr.adilmualadhumad@gmail.com> wrote:
Dear Elaine Meng, Merry Christmas and Happy New Year. When the protein is added to the ligand (as in the attach picture) , the distance between them is remote. How can I get closer to ligand of the future protein. best regards
Adil M. Dhumad
-- Assistant Prof.Dr. Adil Muala Dhumad University of Basrah, College of Education for Pure Sciences, Department of Chemistry
{kind=link}
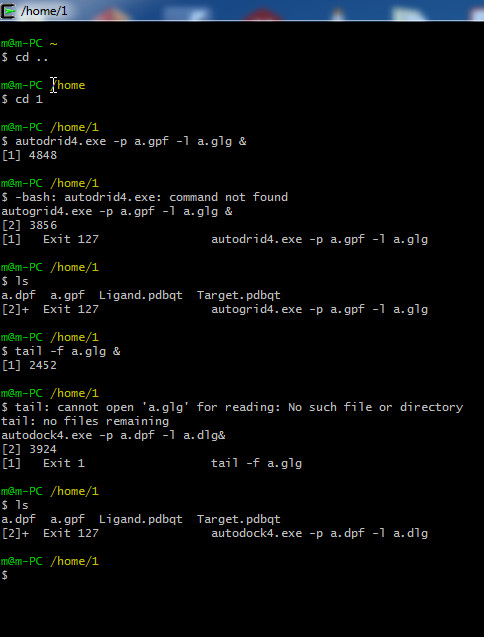
Dear Adil M. Dhumad, This mailing list is for Chimera questions. It looks like you are using Autodock, which is not part of Chimera, and I really have no idea what the problem is. Maybe you can ask the Autodock people. <http://autodock.scripps.edu/> <http://autodock.scripps.edu/wiki/AutoGrid> Also, you really should change the e-mail subject line when you change the subject instead of just reusing it from somebody else’s question, because it is not helpful when people search for answers in the future. Best, Elaine
On Jan 1, 2019, at 5:23 AM, dr. Adil Muala Dhumad Ezirej <dr.adilmualadhumad@gmail.com> wrote:
Dear Elaine Meng, please help me to solution this problem. I'm dont know what's the problem in attach case. Best regards -- Assistant Prof.Dr. Adil Muala Dhumad University of Basrah, College of Education for Pure Sciences, Department of Chemistry <a.jpg>_
Thank you very much! Yangqi On Wednesday, December 26, 2018, Elaine Meng <meng@cgl.ucsf.edu> wrote:
Hi Yangqi, Of course you can open multiple structures and move them how you like and and then save again as PDB or session.
See this previous post for “manual placement of ligand” <http://plato.cgl.ucsf.edu/pipermail/chimera-users/2011-June/006473.html>
Another way is to open your protein pdb file, then open a similar pdb that does have the heme in it, superimpose the two models, and then delete the other protein atoms so that only the heme is left together with your protein. You could just save the PDB file with both models in it, or you could combine them into one model first.
How to superimpose structures: <http://www.rbvi.ucsf.edu/chimera/docs/UsersGuide/superposition.html>
Use menu: Help… Search Documentation to search for “delete” “combine” “save PDB” etc.
I hope this helps, Elaine ----- Elaine C. Meng, Ph.D. UCSF Chimera(X) team Department of Pharmaceutical Chemistry University of California, San Francisco
On Dec 26, 2018, at 12:05 PM, Yangqi Gu <yangqi.gu@yale.edu> wrote:
Dear Chimera users, I have a pdb file of my protein, but not showing the binding ligand (Heme). I know where the binding motif is and I am wondering is there a way I can add ligand to the binding motif and write out the new pdb file in Chimera? Best, Yangqi
-- Yangqi Gu Graduate Student Malvankar Lab Yale University, West Campus
participants (3)
-
 dr. Adil Muala Dhumad Ezirej
dr. Adil Muala Dhumad Ezirej -
 Elaine Meng
Elaine Meng -
 Yangqi Gu
Yangqi Gu