
Hello Elaine, I am using Chmiera-Modeller GUI to add the missing residues for my PDB file: (https://files.rcsb.org/view/1YPH.pdb).
From the mailing list, I am following the procedure listed in: http://www.cgl.ucsf.edu/pipermail/chimera-users/2013-September/009174.html
However, once the missing residues for chains A and B are included, I save the new PDB file using File > Save PDB... > under save models selection "selected all three files" and clicked OK. The resulting PDB file includes the original header with additional 'MODEL 1', 'MODEL 2' and 'MODEL 3'. How to save the atom coordinates as a single PDB file after adding the missing residues? Any comments/suggestions is appreciated. Regards, Daipayan

Dear Daipayan, I do not know what your three models are, because each time you make a model with this tool there is a “number of models” choice. If you chose to only make 1 model for chain A and 1 model for chain B, maybe your three models are: (1) original structure (1yph chains A-F) (2) one model of chain A with missing residues added, now without chain ID (3) one model of chain B with missing residues added, now without chain ID You can see a list of what the current models are by opening the Model Panel (from Favorites menu). I tried opening 1yph (model #0) and making those other models (#1.1 and #2.1) When I hid the original model 1yph (unchecking “S” box in Model Panel) I could see that the new models include not just the peptides, but a bunch of water and sulfate residues (command “display” displays all atoms) If my guess about your three models is correct, I would recommend: (A) deleting the nonprotein parts of the two new models, e.g. command something like: delete #1-2 & ~ protein … meaning delete models #1-2 AND NOT protein (PS be very very careful entering these delete commands because they are irreversible… maybe save session beforehand, just in case) (B) deleting chains A and B from the original structure because you are going to replace them with the new models, e.g. something like: delete #0 & protein & :.a-b (C) merge the 3 models into one, e.g. in Model Panel choose/highlight all three models (rows on the left side) and then use the “copy/combine” button on the right side of the dialog, create new merged model with “rename them uniquely” for any chains that have overlapping IDs … in my test it made the two newly modeled chains G and H instead of A and B … if that bothers you, you can change chain ID (Tools… Structure Editing… Change Chain IDs) (D) then save only the single new merged model, in my case #3, to PDB file An important point is that the Chimera-Modeller interface will only model one chain at a time. It does not care that other chains are there, so any added residues/loops might end up clashing with the other chains. In this case it didn’t seem I hope this helps, Elaine ----- Elaine C. Meng, Ph.D. UCSF Chimera(X) team Department of Pharmaceutical Chemistry University of California, San Francisco
On Nov 14, 2017, at 2:53 AM, Daipayan Sarkar <sdaipayan@gmail.com> wrote:
Hello Elaine,
I am using Chmiera-Modeller GUI to add the missing residues for my PDB file: (https://files.rcsb.org/view/1YPH.pdb).
From the mailing list, I am following the procedure listed in: http://www.cgl.ucsf.edu/pipermail/chimera-users/2013-September/009174.html
However, once the missing residues for chains A and B are included, I save the new PDB file using File > Save PDB... > under save models selection "selected all three files" and clicked OK. The resulting PDB file includes the original header with additional 'MODEL 1', 'MODEL 2' and 'MODEL 3'. How to save the atom coordinates as a single PDB file after adding the missing residues? Any comments/suggestions is appreciated.
Regards, Daipayan
_______________________________________________ Chimera-users mailing list: Chimera-users@cgl.ucsf.edu Manage subscription: http://plato.cgl.ucsf.edu/mailman/listinfo/chimera-users
Forgot to finish this one thought, sorry...
On Nov 14, 2017, at 9:47 AM, Elaine Meng <meng@cgl.ucsf.edu> wrote:
An important point is that the Chimera-Modeller interface will only model one chain at a time. It does not care that other chains are there, so any added residues/loops might end up clashing with the other chains. In this case it didn’t seem
… that bad since the added residues were on the outside of the complex. However, Find Clashes/Contacts (in menu under Tools… Structure Analysis) identified a few clashes between those chains and the other chains in the merged model in my test. I only made one Modeller model of each chain. One reason you might want to try making more than one model of each chain is that they may have different numbers of clashes with the other chains in the structure. However, whether you care about this depends on what you are going to do with this overall structure. If it’s just for a picture, it’s not important, or if you were going to run minimization or dynamics, the clashes would easily resolve themselves in this case because they involve peptide termini on the outside of the complex. Finally, if you want to use Modeller to model the multi-chain complex with awareness of the other chains, you would have to use it separately outside of Chimera. Elaine
Hi Elaine, Thanks for your detailed instructions. My apologies for the confusion but your guess about the different models is correct. After following your instructions, I compared the first residue of the original PDB file with the first residue of 'model 2 (residues missing in chain A)' and following is what I got. Note, the x, y, z coordinates of the atoms have changed and the sequence of atoms has changed. I am not sure why this happens. Can you help me understand? Note, the missing residues where added in model 2 but I am not showing here as there is nothing to compare against. The coordinates for chain A of the first residue in original PDB file: ATOM 1 N CYS A 1 32.449 -7.381 50.051 1.00 18.85 N ATOM 2 CA CYS A 1 31.974 -7.012 48.720 1.00 17.01 C ATOM 3 C CYS A 1 33.101 -6.917 47.728 1.00 15.67 C ATOM 4 O CYS A 1 34.254 -6.671 48.116 1.00 16.39 O ATOM 5 CB CYS A 1 31.253 -5.658 48.764 1.00 17.77 C ATOM 6 SG CYS A 1 32.167 -4.263 49.459 1.00 19.16 S The coordinates for chain A of the first residue in PDB file after adding missing residues using Chimera-Modeller: ATOM 1 N CYS 1 32.426 -7.418 50.092 1.00 0.00 N ATOM 2 CA CYS 1 31.954 -7.042 48.762 1.00 0.00 C ATOM 3 CB CYS 1 31.233 -5.687 48.812 1.00 0.00 C ATOM 4 SG CYS 1 32.146 -4.297 49.517 1.00 0.00 S ATOM 5 C CYS 1 33.082 -6.942 47.773 1.00 0.00 C ATOM 6 O CYS 1 34.235 -6.698 48.164 1.00 0.00 O Regards, Daipayan On Tue, Nov 14, 2017 at 12:00 PM, Elaine Meng <meng@cgl.ucsf.edu> wrote:
Forgot to finish this one thought, sorry...
On Nov 14, 2017, at 9:47 AM, Elaine Meng <meng@cgl.ucsf.edu> wrote:
An important point is that the Chimera-Modeller interface will only model one chain at a time. It does not care that other chains are there, so any added residues/loops might end up clashing with the other chains. In this case it didn’t seem
… that bad since the added residues were on the outside of the complex. However, Find Clashes/Contacts (in menu under Tools… Structure Analysis) identified a few clashes between those chains and the other chains in the merged model in my test. I only made one Modeller model of each chain. One reason you might want to try making more than one model of each chain is that they may have different numbers of clashes with the other chains in the structure. However, whether you care about this depends on what you are going to do with this overall structure. If it’s just for a picture, it’s not important, or if you were going to run minimization or dynamics, the clashes would easily resolve themselves in this case because they involve peptide termini on the outside of the complex.
Finally, if you want to use Modeller to model the multi-chain complex with awareness of the other chains, you would have to use it separately outside of Chimera.
Elaine
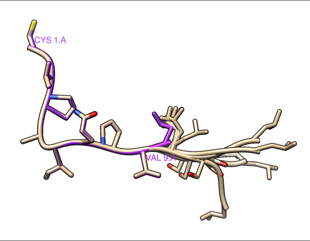
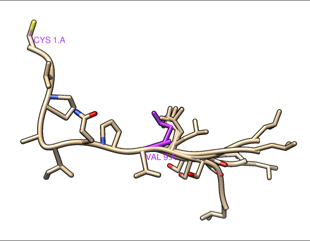
Hi Daipayan, The results are directly from MODELLER, so although I do not know the detailed reasons, it produces small shifts in coordinates and chooses to write the atoms in that order. The shifts appear to be rigid-body movements of the structure as a whole without changing the actual structure (internal coordinates) of the parts that are not being modeled. In other words, if you start with chain A residues 1-10 and make models of chain A residues 1-13 with residues 1-9 kept the same and residue 10 allowed to move, even though the output coordinates of 1-9 are slightly different numbers, you can match them exactly onto the coordinates of 1-9 in the original structure. If you want more detailed reasons you could try asking on the Modeller forum: <https://salilab.org/modeller/discussion_forum.html> As mentioned in my earlier message, the Chimera-Modeller interface only allows modeling 1 chain at a time, so the shifts are completely meaningless as they do not affect the (internal) coordinates of the structure. You might be concerned, however, if you are putting several different modeled chains back together to make a multichain structure. In this case, the shifts are very small, so you could choose to ignore them. Again, it really depends what you are going to do with the resulting final model. If you were really concerned about the small shifts, to get rid of them completely, you could match the model back onto the corresponding atoms of the original structure before saving the coordinates. I tried making 5 models of 1YPH chain A adding the missing C-terminal residues. When I save the models to PDB and then look at the coordinates with a text-editor, they all have similar small shifts of the N-terminal residues as you reported. However, they are barely perceptible when you view the structure. Here the original chain A is purple, the five models tan, without any matching. You can barely see any difference except in the modeled C-terminal part on the right. RMSDs of all 60 atoms of residues 1-9 of my five test models compared to the original were in the range 0.044-0.068. (measured with commands like: rmsd #1:1-9.a #0.1:1-9 …in my case the original is #1 and the five models are #0.1-5) Then I matched the models (#0.1-5) onto the original (#1), commands: match #1:1-9.a #0.5:1-9 match #1:1-9.a #0.4:1-9 match #1:1-9.a #0.3:1-9 match #1:1-9.a #0.2:1-9 match #1:1-9.a #0.1:1-9 … then all the RMSDs were zero (0.001 but that’s within coordinate rounding error). I hope this helps, Elaine ---------- Elaine C. Meng, Ph.D. UCSF Chimera(X) team Department of Pharmaceutical Chemistry University of California, San Francisco
On Nov 15, 2017, at 7:27 PM, Daipayan Sarkar <sdaipayan@gmail.com> wrote:
Hi Elaine,
Thanks for your detailed instructions. My apologies for the confusion but your guess about the different models is correct. After following your instructions, I compared the first residue of the original PDB file with the first residue of 'model 2 (residues missing in chain A)' and following is what I got. Note, the x, y, z coordinates of the atoms have changed and the sequence of atoms has changed. I am not sure why this happens. Can you help me understand? Note, the missing residues where added in model 2 but I am not showing here as there is nothing to compare against.
The coordinates for chain A of the first residue in original PDB file: ATOM 1 N CYS A 1 32.449 -7.381 50.051 1.00 18.85 N ATOM 2 CA CYS A 1 31.974 -7.012 48.720 1.00 17.01 C ATOM 3 C CYS A 1 33.101 -6.917 47.728 1.00 15.67 C ATOM 4 O CYS A 1 34.254 -6.671 48.116 1.00 16.39 O ATOM 5 CB CYS A 1 31.253 -5.658 48.764 1.00 17.77 C ATOM 6 SG CYS A 1 32.167 -4.263 49.459 1.00 19.16 S
The coordinates for chain A of the first residue in PDB file after adding missing residues using Chimera-Modeller: ATOM 1 N CYS 1 32.426 -7.418 50.092 1.00 0.00 N ATOM 2 CA CYS 1 31.954 -7.042 48.762 1.00 0.00 C ATOM 3 CB CYS 1 31.233 -5.687 48.812 1.00 0.00 C ATOM 4 SG CYS 1 32.146 -4.297 49.517 1.00 0.00 S ATOM 5 C CYS 1 33.082 -6.942 47.773 1.00 0.00 C ATOM 6 O CYS 1 34.235 -6.698 48.164 1.00 0.00 O
Regards, Daipayan
On Tue, Nov 14, 2017 at 12:00 PM, Elaine Meng <meng@cgl.ucsf.edu> wrote: Forgot to finish this one thought, sorry...
On Nov 14, 2017, at 9:47 AM, Elaine Meng <meng@cgl.ucsf.edu> wrote:
An important point is that the Chimera-Modeller interface will only model one chain at a time. It does not care that other chains are there, so any added residues/loops might end up clashing with the other chains. In this case it didn’t seem
… that bad since the added residues were on the outside of the complex. However, Find Clashes/Contacts (in menu under Tools… Structure Analysis) identified a few clashes between those chains and the other chains in the merged model in my test. I only made one Modeller model of each chain. One reason you might want to try making more than one model of each chain is that they may have different numbers of clashes with the other chains in the structure. However, whether you care about this depends on what you are going to do with this overall structure. If it’s just for a picture, it’s not important, or if you were going to run minimization or dynamics, the clashes would easily resolve themselves in this case because they involve peptide termini on the outside of the complex.
Finally, if you want to use Modeller to model the multi-chain complex with awareness of the other chains, you would have to use it separately outside of Chimera.
Elaine
{kind=link}
{kind=link}
participants (2)
-
 Daipayan Sarkar
Daipayan Sarkar -
 Elaine Meng
Elaine Meng