
Begin forwarded message:
From: Kenji Matsui <s214903z@st.go.tuat.ac.jp> Subject: Chimera bug report submission Date: December 19, 2021 at 11:33:41 PM PST To: chimera-bugs@cgl.ucsf.edu
The following bug report has been submitted: Platform: windows_x86_64 Chimera Version: 1.16 Description I would like to see the interaction between the ligand and the protein, but I was not able to display it in the way described in the tutorial. How can I do that?
Method select →ligand
Tool Structure analysis Find H-bond Line length 3.0 Å
check Relax H-bond ~, Only find, Include intera-molecular, Include intra-residue
apply →OK File attachment: osh4,OSW-1220.py

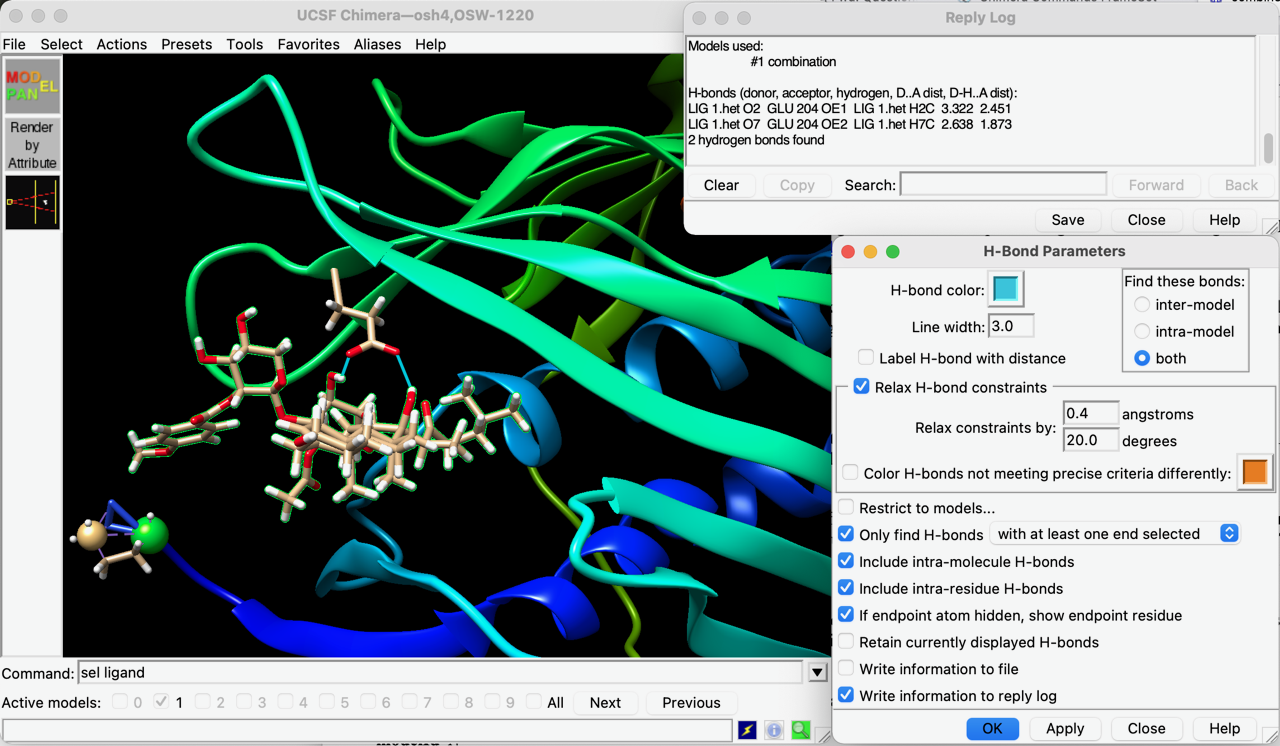
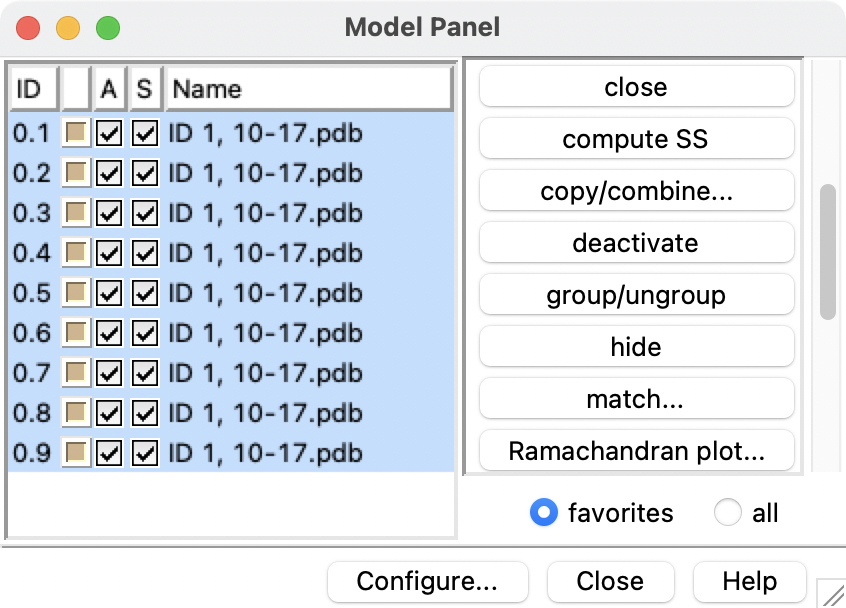
Hi Kenji, This is not a bug. This is because your ligand and receptor molecules are not together in a single model, whereas if you use the same structure that the tutorial is using (or any of many other complexes from the Protein Databank), the ligand and receptor are together in the same model. If you want to follow the tutorial but with your structure, you would need to combine the ligand and receptor into one model. Actually your session contains 10 models, one receptor (#0.1) and 9 ligands (#0.2, #0.3 ... #0.9). I can see this by opening the Model Panel and clicking the "group/ungroup" button on the right, and then using the "S" checkboxes to show and hide each one so that I can see what they contain. Maybe the ligands are all exactly the same as each other, or at least they seem to be quite similar. Here are some example Chimera commands that would combine #0.1 and #0.2 to make a new model #1 and then close all of #0.1-9: combine #0.1,0.2 ref #0.1 model #1 close #0 Now you have one new model #1 with both the receptor (that was originally #0.1) and the ligand (that was originally #0.2). If the other ligands are different, you would need to repeat the combining process for each one if you want to use the same approach to analyze them. Chimera does not automatically recognize this specific ligand as "ligand" (I don't know why), but you can select it by its residue name LIG with menu: Select... Residue... LIG, or command: select :LIG Also, by default FindHBond will automatically find ALL of the H-bonds. It does not care what is selected unless you turn on the option "Only find H-bonds..." e.g. "with at least one end selected". You should also turn on "If endpoint atom hidden, shown endpoint residue" or else it will not show all of the H-bonds it finds. For this ligand, it finds 2 H-bonds as shown in the attached screenshot, with details written to Reply Log. I hope this helps, Elaine ----- Elaine C. Meng, Ph.D. UCSF Chimera(X) team Department of Pharmaceutical Chemistry University of California, San Francisco
On Dec 20, 2021, at 9:07 AM, Eric Pettersen via Chimera-users <chimera-users@cgl.ucsf.edu> wrote:
Begin forwarded message:
From: Kenji Matsui <s214903z@st.go.tuat.ac.jp> Subject: Chimera bug report submission Date: December 19, 2021 at 11:33:41 PM PST To: chimera-bugs@cgl.ucsf.edu
The following bug report has been submitted: Platform: windows_x86_64 Chimera Version: 1.16 Description I would like to see the interaction between the ligand and the protein, but I was not able to display it in the way described in the tutorial. How can I do that?
Method select →ligand
Tool Structure analysis Find H-bond Line length 3.0 Å
check Relax H-bond ~, Only find, Include intera-molecular, Include intra-residue
apply →OK File attachment: osh4,OSW-1220.py <osh4,OSW-1220.py>
{kind=link}
{kind=link}
{kind=link}

Dear Chimera X I apologize for the very late reply. Thank you for your kind reply. It is very helpful that you sent me a screenshot so that I can understand. Thank you for your continued support. --------------------------------------------------------------------------- 松井 健治 東京農工大学大学院 工学府産業技術専攻 修士 〒184-8588 東京都小金井市中町2-24-16 Mail: s214903z@st.go.tuat.ac.jp Kenji Matsui Graduate School of Tokyo University of Agriculture and Technology 2-24-16, Nakamachi, Koganei-shi, Tokyo 184-8588, Japan Mail: s214903z@st.go.tuat.ac.jp 2021年12月21日(火) 4:07 Elaine Meng <meng@cgl.ucsf.edu>:
Hi Kenji, This is not a bug. This is because your ligand and receptor molecules are not together in a single model, whereas if you use the same structure that the tutorial is using (or any of many other complexes from the Protein Databank), the ligand and receptor are together in the same model.
If you want to follow the tutorial but with your structure, you would need to combine the ligand and receptor into one model.
Actually your session contains 10 models, one receptor (#0.1) and 9 ligands (#0.2, #0.3 ... #0.9). I can see this by opening the Model Panel and clicking the "group/ungroup" button on the right, and then using the "S" checkboxes to show and hide each one so that I can see what they contain. Maybe the ligands are all exactly the same as each other, or at least they seem to be quite similar.
Here are some example Chimera commands that would combine #0.1 and #0.2 to make a new model #1 and then close all of #0.1-9:
combine #0.1,0.2 ref #0.1 model #1 close #0
Now you have one new model #1 with both the receptor (that was originally #0.1) and the ligand (that was originally #0.2). If the other ligands are different, you would need to repeat the combining process for each one if you want to use the same approach to analyze them.
Chimera does not automatically recognize this specific ligand as "ligand" (I don't know why), but you can select it by its residue name LIG with menu: Select... Residue... LIG, or command:
select :LIG
Also, by default FindHBond will automatically find ALL of the H-bonds. It does not care what is selected unless you turn on the option "Only find H-bonds..." e.g. "with at least one end selected". You should also turn on "If endpoint atom hidden, shown endpoint residue" or else it will not show all of the H-bonds it finds. For this ligand, it finds 2 H-bonds as shown in the attached screenshot, with details written to Reply Log.
I hope this helps, Elaine ----- Elaine C. Meng, Ph.D. UCSF Chimera(X) team Department of Pharmaceutical Chemistry University of California, San Francisco
On Dec 20, 2021, at 9:07 AM, Eric Pettersen via Chimera-users < chimera-users@cgl.ucsf.edu> wrote:
Begin forwarded message:
From: Kenji Matsui <s214903z@st.go.tuat.ac.jp> Subject: Chimera bug report submission Date: December 19, 2021 at 11:33:41 PM PST To: chimera-bugs@cgl.ucsf.edu
The following bug report has been submitted: Platform: windows_x86_64 Chimera Version: 1.16 Description I would like to see the interaction between the ligand and the protein, but I was not able to display it in the way described in the tutorial. How can I do that?
Method select →ligand
Tool Structure analysis Find H-bond Line length 3.0 Å
check Relax H-bond ~, Only find, Include intera-molecular, Include intra-residue
apply →OK File attachment: osh4,OSW-1220.py
<osh4,OSW-1220.py>
{kind=link}
{kind=link}
{kind=link}
participants (3)
-
 Elaine Meng
Elaine Meng -
 Eric Pettersen
Eric Pettersen -
 Kenji MATSUI
Kenji MATSUI