
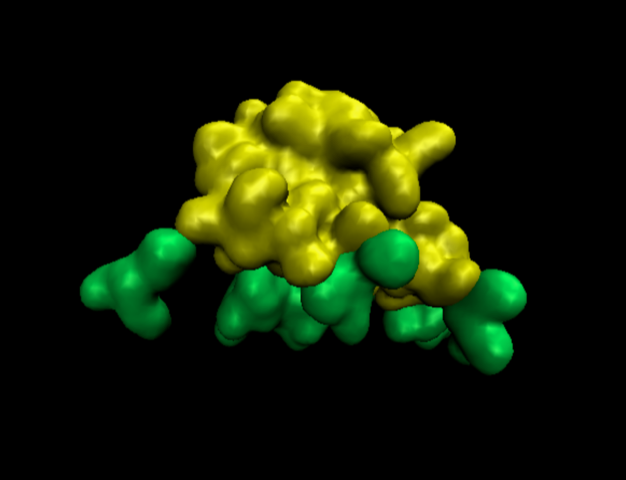
Hello, I would like to know if Chimera could show the surfaces like VMD does with the command QuickSurf (I attach the image with the protein in yellow and the lipids in green). I have been trying with a lot of different variations of the surface command but I can not find the way to make the surface ’softer'…. I have also been trying with spheres representation but the image shows too much information. I need a simplest view, similar to the image because this way it is easier see them move in the .dcd file. It not so important for the protein, just for the lipids. Could you help me with this, please? Thank you very much, clara
{kind=link}

Hi Maria, I see that VMD Quicksurf makes a density map from the atoms and shows an isosurface. You can do the same thing in Chimera with the “molmap” command, but the density map is not automatically recalculated at every step of the trajectory. You would have to use a per-frame script and re-execute molmap at each frame to replace the old map with a new map every time the coordinates change. The molmap command has a resolution parameter and a grid spacing parameter just like Quicksurf. <http://www.rbvi.ucsf.edu/chimera/docs/UsersGuide/midas/molmap.html> You can test by showing one frame of your trajectory and trying various molmap resolution values, e.g. 2.5 angstroms with separate surfaces for protein and non-protein atoms: molmap protein 2.5 grid .5 model 1 molmap ~protein 2.5 grid .5 model 2 Another issue is that the isosurface level isn’t controlled in the molmap command but could be controlled in a separate volume command, e.g.: volume #1 level 0.1 vol #2 level 0.13 If you found commands that worked well enough for you, and you are using MD Movie to view the trajectory, in MD Movie menu: Per-Frame… Define Script and enter these commands as Chimera command script to be executed at each frame. <http://www.rbvi.ucsf.edu/chimera/docs/ContributedSoftware/movie/framemovie.h...> <http://www.rbvi.ucsf.edu/chimera/docs/ContributedSoftware/movie/movie.html#p...> I hope this helps, Elaine ----- Elaine C. Meng, Ph.D. UCSF Chimera(X) team Department of Pharmaceutical Chemistry University of California, San Francisco
On Apr 21, 2019, at 11:57 AM, MARIA CLARA BLANES MIRA <c.blanes@goumh.umh.es> wrote:
Hello, I would like to know if Chimera could show the surfaces like VMD does with the command QuickSurf (I attach the image with the protein in yellow and the lipids in green). I have been trying with a lot of different variations of the surface command but I can not find the way to make the surface ’softer'…. I have also been trying with spheres representation but the image shows too much information. I need a simplest view, similar to the image because this way it is easier see them move in the .dcd file. It not so important for the protein, just for the lipids.
Could you help me with this, please? Thank you very much, clara
Hello Elaine, Just what I needed. Thank you very much. Furthermore, the graphics are great!. The only thing I can’t control is the color because they are changing in a rainbow way when I click on the play in MD Movie. I want the protein always for example blue and the the lipids near the protein always in red. I am trying different options in volume color zones but I can’t find the way. This is what I introduced in the per-frame script: molmap protein 4 grid 0.6 model 1 select :.A z<5 & :DVPC molmap sel 4 grid 0.8 model 2 And it is great for me because they move but it remains the problem with the colours. So, is there any way to fix the colours? Thank you very much for your attention!!, clara
El 22 abr 2019, a las 18:22, Elaine Meng <meng@cgl.ucsf.edu> escribió:
Hi Maria, I see that VMD Quicksurf makes a density map from the atoms and shows an isosurface. You can do the same thing in Chimera with the “molmap” command, but the density map is not automatically recalculated at every step of the trajectory. You would have to use a per-frame script and re-execute molmap at each frame to replace the old map with a new map every time the coordinates change.
The molmap command has a resolution parameter and a grid spacing parameter just like Quicksurf. <http://www.rbvi.ucsf.edu/chimera/docs/UsersGuide/midas/molmap.html>
You can test by showing one frame of your trajectory and trying various molmap resolution values, e.g. 2.5 angstroms with separate surfaces for protein and non-protein atoms:
molmap protein 2.5 grid .5 model 1 molmap ~protein 2.5 grid .5 model 2
Another issue is that the isosurface level isn’t controlled in the molmap command but could be controlled in a separate volume command, e.g.:
volume #1 level 0.1 vol #2 level 0.13
If you found commands that worked well enough for you, and you are using MD Movie to view the trajectory, in MD Movie menu: Per-Frame… Define Script and enter these commands as Chimera command script to be executed at each frame. <http://www.rbvi.ucsf.edu/chimera/docs/ContributedSoftware/movie/framemovie.h...> <http://www.rbvi.ucsf.edu/chimera/docs/ContributedSoftware/movie/movie.html#p...>
I hope this helps, Elaine ----- Elaine C. Meng, Ph.D. UCSF Chimera(X) team Department of Pharmaceutical Chemistry University of California, San Francisco
On Apr 21, 2019, at 11:57 AM, MARIA CLARA BLANES MIRA <c.blanes@goumh.umh.es> wrote:
Hello, I would like to know if Chimera could show the surfaces like VMD does with the command QuickSurf (I attach the image with the protein in yellow and the lipids in green). I have been trying with a lot of different variations of the surface command but I can not find the way to make the surface ’softer'…. I have also been trying with spheres representation but the image shows too much information. I need a simplest view, similar to the image because this way it is easier see them move in the .dcd file. It not so important for the protein, just for the lipids.
Could you help me with this, please? Thank you very much, clara
Hi Clara (oops sorry for using the wrong name before), You can include coloring in the script, e.g. volume #1 color blue volume #2 color red Also to avoid selecting at every frame, you may be able to specify atoms directly in the molmap command, e.g. molmap ":.A z<5 & :DVPC” 4 grid 0.8 model 2 The quotation marks should be plain-text quotation marks, not the fancy curved ones that may be shown in this message because the Mail app changes them automatically. I hope this helps, Elaine ----- Elaine C. Meng, Ph.D. UCSF Chimera(X) team Department of Pharmaceutical Chemistry University of California, San Francisco
On Apr 22, 2019, at 10:43 AM, MARIA CLARA BLANES MIRA <c.blanes@goumh.umh.es> wrote:
Hello Elaine, Just what I needed. Thank you very much. Furthermore, the graphics are great!.
The only thing I can’t control is the color because they are changing in a rainbow way when I click on the play in MD Movie. I want the protein always for example blue and the the lipids near the protein always in red. I am trying different options in volume color zones but I can’t find the way. This is what I introduced in the per-frame script:
molmap protein 4 grid 0.6 model 1 select :.A z<5 & :DVPC molmap sel 4 grid 0.8 model 2
And it is great for me because they move but it remains the problem with the colours. So, is there any way to fix the colours? Thank you very much for your attention!!, clara
El 22 abr 2019, a las 18:22, Elaine Meng <meng@cgl.ucsf.edu> escribió:
Hi Maria, I see that VMD Quicksurf makes a density map from the atoms and shows an isosurface. You can do the same thing in Chimera with the “molmap” command, but the density map is not automatically recalculated at every step of the trajectory. You would have to use a per-frame script and re-execute molmap at each frame to replace the old map with a new map every time the coordinates change.
The molmap command has a resolution parameter and a grid spacing parameter just like Quicksurf. <http://www.rbvi.ucsf.edu/chimera/docs/UsersGuide/midas/molmap.html>
You can test by showing one frame of your trajectory and trying various molmap resolution values, e.g. 2.5 angstroms with separate surfaces for protein and non-protein atoms:
molmap protein 2.5 grid .5 model 1 molmap ~protein 2.5 grid .5 model 2
Another issue is that the isosurface level isn’t controlled in the molmap command but could be controlled in a separate volume command, e.g.:
volume #1 level 0.1 vol #2 level 0.13
If you found commands that worked well enough for you, and you are using MD Movie to view the trajectory, in MD Movie menu: Per-Frame… Define Script and enter these commands as Chimera command script to be executed at each frame. <http://www.rbvi.ucsf.edu/chimera/docs/ContributedSoftware/movie/framemovie.h...> <http://www.rbvi.ucsf.edu/chimera/docs/ContributedSoftware/movie/movie.html#p...>
I hope this helps, Elaine ----- Elaine C. Meng, Ph.D. UCSF Chimera(X) team Department of Pharmaceutical Chemistry University of California, San Francisco
On Apr 21, 2019, at 11:57 AM, MARIA CLARA BLANES MIRA <c.blanes@goumh.umh.es> wrote:
Hello, I would like to know if Chimera could show the surfaces like VMD does with the command QuickSurf (I attach the image with the protein in yellow and the lipids in green). I have been trying with a lot of different variations of the surface command but I can not find the way to make the surface ’softer'…. I have also been trying with spheres representation but the image shows too much information. I need a simplest view, similar to the image because this way it is easier see them move in the .dcd file. It not so important for the protein, just for the lipids.
Could you help me with this, please? Thank you very much, clara
Dear Elaine, Thank you very much for your suggestions and help. It is perfect now! Best regards, Clara
El 22 abr 2019, a las 23:15, Elaine Meng <meng@cgl.ucsf.edu> escribió:
Hi Clara (oops sorry for using the wrong name before),
You can include coloring in the script, e.g.
volume #1 color blue volume #2 color red
Also to avoid selecting at every frame, you may be able to specify atoms directly in the molmap command, e.g.
molmap ":.A z<5 & :DVPC” 4 grid 0.8 model 2
The quotation marks should be plain-text quotation marks, not the fancy curved ones that may be shown in this message because the Mail app changes them automatically.
I hope this helps, Elaine ----- Elaine C. Meng, Ph.D. UCSF Chimera(X) team Department of Pharmaceutical Chemistry University of California, San Francisco
On Apr 22, 2019, at 10:43 AM, MARIA CLARA BLANES MIRA <c.blanes@goumh.umh.es> wrote:
Hello Elaine, Just what I needed. Thank you very much. Furthermore, the graphics are great!.
The only thing I can’t control is the color because they are changing in a rainbow way when I click on the play in MD Movie. I want the protein always for example blue and the the lipids near the protein always in red. I am trying different options in volume color zones but I can’t find the way. This is what I introduced in the per-frame script:
molmap protein 4 grid 0.6 model 1 select :.A z<5 & :DVPC molmap sel 4 grid 0.8 model 2
And it is great for me because they move but it remains the problem with the colours. So, is there any way to fix the colours? Thank you very much for your attention!!, clara
El 22 abr 2019, a las 18:22, Elaine Meng <meng@cgl.ucsf.edu> escribió:
Hi Maria, I see that VMD Quicksurf makes a density map from the atoms and shows an isosurface. You can do the same thing in Chimera with the “molmap” command, but the density map is not automatically recalculated at every step of the trajectory. You would have to use a per-frame script and re-execute molmap at each frame to replace the old map with a new map every time the coordinates change.
The molmap command has a resolution parameter and a grid spacing parameter just like Quicksurf. <http://www.rbvi.ucsf.edu/chimera/docs/UsersGuide/midas/molmap.html>
You can test by showing one frame of your trajectory and trying various molmap resolution values, e.g. 2.5 angstroms with separate surfaces for protein and non-protein atoms:
molmap protein 2.5 grid .5 model 1 molmap ~protein 2.5 grid .5 model 2
Another issue is that the isosurface level isn’t controlled in the molmap command but could be controlled in a separate volume command, e.g.:
volume #1 level 0.1 vol #2 level 0.13
If you found commands that worked well enough for you, and you are using MD Movie to view the trajectory, in MD Movie menu: Per-Frame… Define Script and enter these commands as Chimera command script to be executed at each frame. <http://www.rbvi.ucsf.edu/chimera/docs/ContributedSoftware/movie/framemovie.h...> <http://www.rbvi.ucsf.edu/chimera/docs/ContributedSoftware/movie/movie.html#p...>
I hope this helps, Elaine ----- Elaine C. Meng, Ph.D. UCSF Chimera(X) team Department of Pharmaceutical Chemistry University of California, San Francisco
On Apr 21, 2019, at 11:57 AM, MARIA CLARA BLANES MIRA <c.blanes@goumh.umh.es> wrote:
Hello, I would like to know if Chimera could show the surfaces like VMD does with the command QuickSurf (I attach the image with the protein in yellow and the lipids in green). I have been trying with a lot of different variations of the surface command but I can not find the way to make the surface ’softer'…. I have also been trying with spheres representation but the image shows too much information. I need a simplest view, similar to the image because this way it is easier see them move in the .dcd file. It not so important for the protein, just for the lipids.
Could you help me with this, please? Thank you very much, clara
Dear Elaine, I'm trying to go a little further with the isosurfaces case… I would like to make a movie showing the movements of some lipids when they interact with the protein. The way you suggested me before works perfectly but now I would like to incorporate the perframe script commands into the first script that I designed, the .com script. And here is where I find find the problem because I can’t move the protein and the lipids as I did with the MD movie window. I am trying to combine both commands, perframe and coordset (I think this is the way to do it with commands) but I only obtain errors. Is it possible to do the protein and lipids movement part of the movie with commands instead of the MD movie windows? Here is the .com script: #protein in isosurface rep movie record molmap protein 4 grid 0.6 model 1 volume #1 color #dae4bceb8d98 wait ~ribbon :.a movie crossfade 60 #PC and PA phosphates in sphere rep repr sphere :DVPC & P color #aaaa84bdc71c :DVPC & P movie crossfade 30 2dlabel create l1 text 'Phosphatidylcholine (PC)' color #84bd4bdaaaaa ypos 0.9 xpos .7 size 68 typeface serif wait 30 repr sphere :DVPA & P movie crossfade 30 color #aaaab425684b :DVPA & P movie crossfade 30 2dlabel create l2 text 'Phosphatidic Acid (PA)' color #4bda71c725ed ypos 0.9 xpos .7 size 68 typeface serif movie crossfade 30 #hide tails ~disp :DVPC & C ~disp :DVPC & H ~disp :DVPC & O ~disp :DVPC & N ~disp :DVPA & C ~disp :DVPA & H ~disp :DVPA & O #select DVPC and DVPA at z > 5 from the protein to make them transparent select :.A z > 5 & :DVPC transp 50,a :DVPC select :.A z > 5 & :DVPA transp 50,a :DVPA movie crossfade 30 2dlabel delete * #select DVPC and DVPA lipids at z < 5 from the protein to create isosurfaces molmap ":.A z<5 & :DVPC" 4 grid 0.8 model 2 ~ disp element.H volume #2 color #aaaa84bdc71c movie crossfade 100 molmap ":.A z<5 & :DVPA" 4 grid 0.8 model 3 ~ disp element.H volume #3 color #aaaab425684b At this point this is the representation that I obtain: And now it is the time for the trajectories movements... What I have been trying is: perframe coordset #1-3 20,50,1 or perframe "coordset #1-3 20,50,1” and other different variants but I only obtain errors. Could you help me with this issue, please?, clara
El 22 abr 2019, a las 23:15, Elaine Meng <meng@cgl.ucsf.edu> escribió:
Hi Clara (oops sorry for using the wrong name before),
You can include coloring in the script, e.g.
volume #1 color blue volume #2 color red
Also to avoid selecting at every frame, you may be able to specify atoms directly in the molmap command, e.g.
molmap ":.A z<5 & :DVPC” 4 grid 0.8 model 2
The quotation marks should be plain-text quotation marks, not the fancy curved ones that may be shown in this message because the Mail app changes them automatically.
I hope this helps, Elaine ----- Elaine C. Meng, Ph.D. UCSF Chimera(X) team Department of Pharmaceutical Chemistry University of California, San Francisco
On Apr 22, 2019, at 10:43 AM, MARIA CLARA BLANES MIRA <c.blanes@goumh.umh.es> wrote:
Hello Elaine, Just what I needed. Thank you very much. Furthermore, the graphics are great!.
The only thing I can’t control is the color because they are changing in a rainbow way when I click on the play in MD Movie. I want the protein always for example blue and the the lipids near the protein always in red. I am trying different options in volume color zones but I can’t find the way. This is what I introduced in the per-frame script:
molmap protein 4 grid 0.6 model 1 select :.A z<5 & :DVPC molmap sel 4 grid 0.8 model 2
And it is great for me because they move but it remains the problem with the colours. So, is there any way to fix the colours? Thank you very much for your attention!!, clara
El 22 abr 2019, a las 18:22, Elaine Meng <meng@cgl.ucsf.edu> escribió:
Hi Maria, I see that VMD Quicksurf makes a density map from the atoms and shows an isosurface. You can do the same thing in Chimera with the “molmap” command, but the density map is not automatically recalculated at every step of the trajectory. You would have to use a per-frame script and re-execute molmap at each frame to replace the old map with a new map every time the coordinates change.
The molmap command has a resolution parameter and a grid spacing parameter just like Quicksurf. <http://www.rbvi.ucsf.edu/chimera/docs/UsersGuide/midas/molmap.html>
You can test by showing one frame of your trajectory and trying various molmap resolution values, e.g. 2.5 angstroms with separate surfaces for protein and non-protein atoms:
molmap protein 2.5 grid .5 model 1 molmap ~protein 2.5 grid .5 model 2
Another issue is that the isosurface level isn’t controlled in the molmap command but could be controlled in a separate volume command, e.g.:
volume #1 level 0.1 vol #2 level 0.13
If you found commands that worked well enough for you, and you are using MD Movie to view the trajectory, in MD Movie menu: Per-Frame… Define Script and enter these commands as Chimera command script to be executed at each frame. <http://www.rbvi.ucsf.edu/chimera/docs/ContributedSoftware/movie/framemovie.h...> <http://www.rbvi.ucsf.edu/chimera/docs/ContributedSoftware/movie/movie.html#p...>
I hope this helps, Elaine ----- Elaine C. Meng, Ph.D. UCSF Chimera(X) team Department of Pharmaceutical Chemistry University of California, San Francisco
On Apr 21, 2019, at 11:57 AM, MARIA CLARA BLANES MIRA <c.blanes@goumh.umh.es> wrote:
Hello, I would like to know if Chimera could show the surfaces like VMD does with the command QuickSurf (I attach the image with the protein in yellow and the lipids in green). I have been trying with a lot of different variations of the surface command but I can not find the way to make the surface ’softer'…. I have also been trying with spheres representation but the image shows too much information. I need a simplest view, similar to the image because this way it is easier see them move in the .dcd file. It not so important for the protein, just for the lipids.
Could you help me with this, please? Thank you very much, clara
{kind=link}
Hi Clara, The perframe command is used to specify what should happen at every frame, just like the per-frame script in MD Movie. The coordset command shows multiple frames of the trajectory (like using the play button in MD Movie), so it doesn’t make sense to have it as part of the perframe actions. So if your per-frame script in MD Movie had command1 command2 command3 Then you could define an alias for the perframe actions like this “alias” command, where multiple commands are separated by semicolons: alias ^myactions command1; command2; command3 <http://www.rbvi.ucsf.edu/chimera/docs/UsersGuide/midas/alias.html> ...(substituting the actual commands, of course, and “myactions” could be some other word you want to name the alias) …and then in your overall script where you want to show the trajectory, start running the actions with perframe, use the coordset command to play the frames of the atomic trajectory, and after waiting the same number of frames as you played with coordset, turn off the perframe actions. Example command to do all this is below. Coordset is not used for volumes. It shows only the atomic trajectory (again like pressing the play button in MD Movie), and your per-frame actions create the volume models from the atoms and color them just as we discussed earlier. If your atomic trajectory is #1, then the command could be something like perframe myactions; coordset #1 20,50,1; wait 31; ~perframe I hope this helps, Elaine ----- Elaine C. Meng, Ph.D. UCSF Chimera(X) team Department of Pharmaceutical Chemistry University of California, San Francisco
On Apr 24, 2019, at 10:36 AM, MARIA CLARA BLANES MIRA <c.blanes@goumh.umh.es> wrote:
Dear Elaine, I'm trying to go a little further with the isosurfaces case… I would like to make a movie showing the movements of some lipids when they interact with the protein. The way you suggested me before works perfectly but now I would like to incorporate the perframe script commands into the first script that I designed, the .com script. And here is where I find find the problem because I can’t move the protein and the lipids as I did with the MD movie window. I am trying to combine both commands, perframe and coordset (I think this is the way to do it with commands) but I only obtain errors. Is it possible to do the protein and lipids movement part of the movie with commands instead of the MD movie windows?
Here is the .com script:
#protein in isosurface rep movie record molmap protein 4 grid 0.6 model 1 volume #1 color #dae4bceb8d98 wait ~ribbon :.a movie crossfade 60
#PC and PA phosphates in sphere rep repr sphere :DVPC & P color #aaaa84bdc71c :DVPC & P movie crossfade 30 2dlabel create l1 text 'Phosphatidylcholine (PC)' color #84bd4bdaaaaa ypos 0.9 xpos .7 size 68 typeface serif wait 30
repr sphere :DVPA & P movie crossfade 30 color #aaaab425684b :DVPA & P movie crossfade 30 2dlabel create l2 text 'Phosphatidic Acid (PA)' color #4bda71c725ed ypos 0.9 xpos .7 size 68 typeface serif movie crossfade 30
#hide tails ~disp :DVPC & C ~disp :DVPC & H ~disp :DVPC & O ~disp :DVPC & N
~disp :DVPA & C ~disp :DVPA & H ~disp :DVPA & O
#select DVPC and DVPA at z > 5 from the protein to make them transparent select :.A z > 5 & :DVPC transp 50,a :DVPC select :.A z > 5 & :DVPA transp 50,a :DVPA movie crossfade 30
2dlabel delete *
#select DVPC and DVPA lipids at z < 5 from the protein to create isosurfaces molmap ":.A z<5 & :DVPC" 4 grid 0.8 model 2 ~ disp element.H volume #2 color #aaaa84bdc71c movie crossfade 100
molmap ":.A z<5 & :DVPA" 4 grid 0.8 model 3 ~ disp element.H volume #3 color #aaaab425684b
At this point this is the representation that I obtain:
<Captura de pantalla 2019-04-24 a las 18.52.56.png>
And now it is the time for the trajectories movements... What I have been trying is: perframe coordset #1-3 20,50,1 or perframe "coordset #1-3 20,50,1” and other different variants but I only obtain errors.
Could you help me with this issue, please?, clara
El 22 abr 2019, a las 23:15, Elaine Meng <meng@cgl.ucsf.edu> escribió:
Hi Clara (oops sorry for using the wrong name before),
You can include coloring in the script, e.g.
volume #1 color blue volume #2 color red
Also to avoid selecting at every frame, you may be able to specify atoms directly in the molmap command, e.g.
molmap ":.A z<5 & :DVPC” 4 grid 0.8 model 2
The quotation marks should be plain-text quotation marks, not the fancy curved ones that may be shown in this message because the Mail app changes them automatically.
I hope this helps, Elaine ----- Elaine C. Meng, Ph.D. UCSF Chimera(X) team Department of Pharmaceutical Chemistry University of California, San Francisco
On Apr 22, 2019, at 10:43 AM, MARIA CLARA BLANES MIRA <c.blanes@goumh.umh.es> wrote:
Hello Elaine, Just what I needed. Thank you very much. Furthermore, the graphics are great!.
The only thing I can’t control is the color because they are changing in a rainbow way when I click on the play in MD Movie. I want the protein always for example blue and the the lipids near the protein always in red. I am trying different options in volume color zones but I can’t find the way. This is what I introduced in the per-frame script:
molmap protein 4 grid 0.6 model 1 select :.A z<5 & :DVPC molmap sel 4 grid 0.8 model 2
And it is great for me because they move but it remains the problem with the colours. So, is there any way to fix the colours? Thank you very much for your attention!!, clara
El 22 abr 2019, a las 18:22, Elaine Meng <meng@cgl.ucsf.edu> escribió:
Hi Maria, I see that VMD Quicksurf makes a density map from the atoms and shows an isosurface. You can do the same thing in Chimera with the “molmap” command, but the density map is not automatically recalculated at every step of the trajectory. You would have to use a per-frame script and re-execute molmap at each frame to replace the old map with a new map every time the coordinates change.
The molmap command has a resolution parameter and a grid spacing parameter just like Quicksurf. <http://www.rbvi.ucsf.edu/chimera/docs/UsersGuide/midas/molmap.html>
You can test by showing one frame of your trajectory and trying various molmap resolution values, e.g. 2.5 angstroms with separate surfaces for protein and non-protein atoms:
molmap protein 2.5 grid .5 model 1 molmap ~protein 2.5 grid .5 model 2
Another issue is that the isosurface level isn’t controlled in the molmap command but could be controlled in a separate volume command, e.g.:
volume #1 level 0.1 vol #2 level 0.13
If you found commands that worked well enough for you, and you are using MD Movie to view the trajectory, in MD Movie menu: Per-Frame… Define Script and enter these commands as Chimera command script to be executed at each frame. <http://www.rbvi.ucsf.edu/chimera/docs/ContributedSoftware/movie/framemovie.h...> <http://www.rbvi.ucsf.edu/chimera/docs/ContributedSoftware/movie/movie.html#p...>
I hope this helps, Elaine
Thank you very much Elaine, Best regards, Clara
El 24 abr 2019, a las 23:07, Elaine Meng <meng@cgl.ucsf.edu> escribió:
Hi Clara, The perframe command is used to specify what should happen at every frame, just like the per-frame script in MD Movie. The coordset command shows multiple frames of the trajectory (like using the play button in MD Movie), so it doesn’t make sense to have it as part of the perframe actions. So if your per-frame script in MD Movie had
command1 command2 command3
Then you could define an alias for the perframe actions like this “alias” command, where multiple commands are separated by semicolons:
alias ^myactions command1; command2; command3
<http://www.rbvi.ucsf.edu/chimera/docs/UsersGuide/midas/alias.html>
...(substituting the actual commands, of course, and “myactions” could be some other word you want to name the alias) …and then in your overall script where you want to show the trajectory, start running the actions with perframe, use the coordset command to play the frames of the atomic trajectory, and after waiting the same number of frames as you played with coordset, turn off the perframe actions. Example command to do all this is below.
Coordset is not used for volumes. It shows only the atomic trajectory (again like pressing the play button in MD Movie), and your per-frame actions create the volume models from the atoms and color them just as we discussed earlier. If your atomic trajectory is #1, then the command could be something like
perframe myactions; coordset #1 20,50,1; wait 31; ~perframe
I hope this helps, Elaine ----- Elaine C. Meng, Ph.D. UCSF Chimera(X) team Department of Pharmaceutical Chemistry University of California, San Francisco
On Apr 24, 2019, at 10:36 AM, MARIA CLARA BLANES MIRA <c.blanes@goumh.umh.es> wrote:
Dear Elaine, I'm trying to go a little further with the isosurfaces case… I would like to make a movie showing the movements of some lipids when they interact with the protein. The way you suggested me before works perfectly but now I would like to incorporate the perframe script commands into the first script that I designed, the .com script. And here is where I find find the problem because I can’t move the protein and the lipids as I did with the MD movie window. I am trying to combine both commands, perframe and coordset (I think this is the way to do it with commands) but I only obtain errors. Is it possible to do the protein and lipids movement part of the movie with commands instead of the MD movie windows?
Here is the .com script:
#protein in isosurface rep movie record molmap protein 4 grid 0.6 model 1 volume #1 color #dae4bceb8d98 wait ~ribbon :.a movie crossfade 60
#PC and PA phosphates in sphere rep repr sphere :DVPC & P color #aaaa84bdc71c :DVPC & P movie crossfade 30 2dlabel create l1 text 'Phosphatidylcholine (PC)' color #84bd4bdaaaaa ypos 0.9 xpos .7 size 68 typeface serif wait 30
repr sphere :DVPA & P movie crossfade 30 color #aaaab425684b :DVPA & P movie crossfade 30 2dlabel create l2 text 'Phosphatidic Acid (PA)' color #4bda71c725ed ypos 0.9 xpos .7 size 68 typeface serif movie crossfade 30
#hide tails ~disp :DVPC & C ~disp :DVPC & H ~disp :DVPC & O ~disp :DVPC & N
~disp :DVPA & C ~disp :DVPA & H ~disp :DVPA & O
#select DVPC and DVPA at z > 5 from the protein to make them transparent select :.A z > 5 & :DVPC transp 50,a :DVPC select :.A z > 5 & :DVPA transp 50,a :DVPA movie crossfade 30
2dlabel delete *
#select DVPC and DVPA lipids at z < 5 from the protein to create isosurfaces molmap ":.A z<5 & :DVPC" 4 grid 0.8 model 2 ~ disp element.H volume #2 color #aaaa84bdc71c movie crossfade 100
molmap ":.A z<5 & :DVPA" 4 grid 0.8 model 3 ~ disp element.H volume #3 color #aaaab425684b
At this point this is the representation that I obtain:
<Captura de pantalla 2019-04-24 a las 18.52.56.png>
And now it is the time for the trajectories movements... What I have been trying is: perframe coordset #1-3 20,50,1 or perframe "coordset #1-3 20,50,1” and other different variants but I only obtain errors.
Could you help me with this issue, please?, clara
El 22 abr 2019, a las 23:15, Elaine Meng <meng@cgl.ucsf.edu> escribió:
Hi Clara (oops sorry for using the wrong name before),
You can include coloring in the script, e.g.
volume #1 color blue volume #2 color red
Also to avoid selecting at every frame, you may be able to specify atoms directly in the molmap command, e.g.
molmap ":.A z<5 & :DVPC” 4 grid 0.8 model 2
The quotation marks should be plain-text quotation marks, not the fancy curved ones that may be shown in this message because the Mail app changes them automatically.
I hope this helps, Elaine ----- Elaine C. Meng, Ph.D. UCSF Chimera(X) team Department of Pharmaceutical Chemistry University of California, San Francisco
On Apr 22, 2019, at 10:43 AM, MARIA CLARA BLANES MIRA <c.blanes@goumh.umh.es> wrote:
Hello Elaine, Just what I needed. Thank you very much. Furthermore, the graphics are great!.
The only thing I can’t control is the color because they are changing in a rainbow way when I click on the play in MD Movie. I want the protein always for example blue and the the lipids near the protein always in red. I am trying different options in volume color zones but I can’t find the way. This is what I introduced in the per-frame script:
molmap protein 4 grid 0.6 model 1 select :.A z<5 & :DVPC molmap sel 4 grid 0.8 model 2
And it is great for me because they move but it remains the problem with the colours. So, is there any way to fix the colours? Thank you very much for your attention!!, clara
El 22 abr 2019, a las 18:22, Elaine Meng <meng@cgl.ucsf.edu> escribió:
Hi Maria, I see that VMD Quicksurf makes a density map from the atoms and shows an isosurface. You can do the same thing in Chimera with the “molmap” command, but the density map is not automatically recalculated at every step of the trajectory. You would have to use a per-frame script and re-execute molmap at each frame to replace the old map with a new map every time the coordinates change.
The molmap command has a resolution parameter and a grid spacing parameter just like Quicksurf. <http://www.rbvi.ucsf.edu/chimera/docs/UsersGuide/midas/molmap.html>
You can test by showing one frame of your trajectory and trying various molmap resolution values, e.g. 2.5 angstroms with separate surfaces for protein and non-protein atoms:
molmap protein 2.5 grid .5 model 1 molmap ~protein 2.5 grid .5 model 2
Another issue is that the isosurface level isn’t controlled in the molmap command but could be controlled in a separate volume command, e.g.:
volume #1 level 0.1 vol #2 level 0.13
If you found commands that worked well enough for you, and you are using MD Movie to view the trajectory, in MD Movie menu: Per-Frame… Define Script and enter these commands as Chimera command script to be executed at each frame. <http://www.rbvi.ucsf.edu/chimera/docs/ContributedSoftware/movie/framemovie.h...> <http://www.rbvi.ucsf.edu/chimera/docs/ContributedSoftware/movie/movie.html#p...>
I hope this helps, Elaine
participants (2)
-
 Elaine Meng
Elaine Meng -
 MARIA CLARA BLANES MIRA
MARIA CLARA BLANES MIRA