
Dear sir or madam, I am a recent user of the chimera software on Linux-Ubuntu (11.10) operating system and I confronted a dead end on the following issue. I reconstructed a missing side chain of 25 amino acids in a PDB file but I cant seem to find how to set the secondary structure and so on for these 25 residues. So they are stuck in a linear formation externaly from my protein. For your consideration I am attaching also the PDB file. Thank you in advance and looking forward in hearing from you regarding my problem. Best Regards, Sotirios Katsamakas

Dear Sotirios Katsamakas, You didn't say how you added the 25 amino acids. If I were adding them in Chimera, I would: (1) first build a peptide of the 25 amino acids as a separate model, using Build Structure (under Tools... Structure Editing), Start Structure section, "peptide sequence" -- this lets you specify all the phi and psi angles before the peptide is created <http://www.cgl.ucsf.edu/chimera/docs/ContributedSoftware/editing/editing.htm...> (2) join the original protein and the new peptide using Build Structure, Join Models section <http://www.cgl.ucsf.edu/chimera/docs/ContributedSoftware/editing/editing.htm...> So one possibility is to delete the 25 amino acids from that structure and build them again in Chimera as described above. A second possibility is to rotate each phi and psi angle in your current structure, using either Build Structure, Adjust Torsions section, or the "rotation" command. However, I think that would be difficult, or at least quite tedious and labor-intensive. <http://www.cgl.ucsf.edu/chimera/docs/ContributedSoftware/editing/editing.htm...> <http://www.cgl.ucsf.edu/chimera/docs/UsersGuide/midas/rotation.html> I hope this helps, Elaine ---------- Elaine C. Meng, Ph.D. UCSF Computer Graphics Lab (Chimera team) and Babbitt Lab Department of Pharmaceutical Chemistry University of California, San Francisco On Sep 13, 2012, at 5:40 AM, Sotirios Katsamakas wrote:
Dear sir or madam, I am a recent user of the chimera software on Linux-Ubuntu (11.10) operating system and I confronted a dead end on the following issue. I reconstructed a missing side chain of 25 amino acids in a PDB file but I cant seem to find how to set the secondary structure and so on for these 25 residues. So they are stuck in a linear formation externaly from my protein. For your consideration I am attaching also the PDB file. Thank you in advance and looking forward in hearing from you regarding my problem.
Best Regards, Sotirios Katsamakas
<1HVY-reconstructed_pdbv3.pdb>

On Sep 13, 2012, at 9:17 AM, Elaine Meng wrote:
A second possibility is to rotate each phi and psi angle in your current structure, using either Build Structure, Adjust Torsions section, or the "rotation" command. However, I think that would be difficult, or at least quite tedious and labor-intensive.
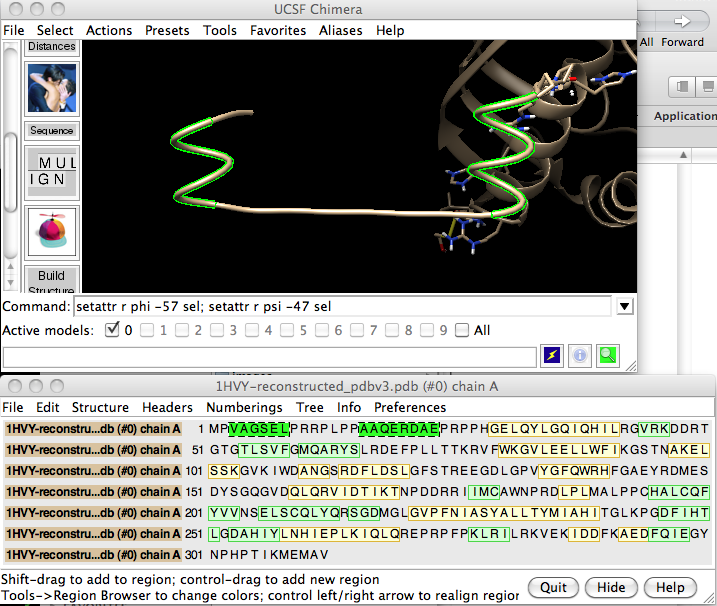
Along these same lines, since phi/psi angles are residue attributes, you can use the setattr command to set them. For instance, an alpha helix has phi/psi angles of -57/-47. So your original extended structure that looked like this: Could at least be changed into a couple of alpha helical segments like so: Your extended structure has a lot of prolines in it, which means it cannot easily be made into one helix since prolines cannot have their phi angles changed without corresponding adjustments to the side chain ring (which is why prolines are known as "helix breaking" residues), so maybe the other approaches that Elaine suggested are better here. --Eric
{kind=link}
{kind=link}
participants (3)
-
 Elaine Meng
Elaine Meng -
 Eric Pettersen
Eric Pettersen -
 Sotirios Katsamakas
Sotirios Katsamakas