Re: [Chimera-users] SURF for large proteins

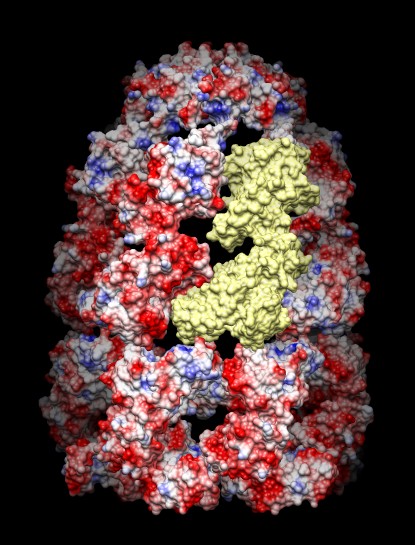
Hi Vijay, Chimera molecular surface calculation (using MSMS) fails for most molecules larger than 5,000 atoms. The main trick that can help is to split large multi-chain models into a separate model for each chain using the split command (e.g. "split #0"). Then use a separate molecular surface on each chain. The attached photo shows the result of that for chaperone 1aon with 21 chains with one subunit colored yellow so you can see its shape. If you use Coulombic Electrostatic Coloring (menu Tools / Surface & Binding Analysis) doing it on the split model gives a slightly different result than with an unsplit model. The potential on each surface is just the potential for one chain. Doing the calculation without split would use the potential for the entire model. The split version can be more useful if you are interested in complementary charges on interfaces between chains. Sometimes you can get a molecular surface computation that fails on one computer to work on another computer. Mac Chimera produces surfaces for larger models, and Windows Chimera has the most failures, and the failure rate for 64-bit Windows Chimera is much higher than 32-bit Windows Chimera which is higher than Mac (all versions). Not sure where Linux fits in. Another approach is to use a different kind of surface, one generated from a density map made by placing Gaussians at each atom. The molmap command creates this surface (e.g. "molmap #0 5" for a 5 Angstrom resolution surface). You'll probably want to use a high resolution (< 5A) since the electrostatics can vary on short distances. You might want to use the molmap "gridSpacing" option to get a finer mesh for the surface (e.g. "molmap #0 5 gridSpacing 1" for a 1 Angstrom grid). Currently only molecular surfaces can be colored by Chimera's Coulomb potential coloring code, so that won't work on these molmap surfaces. You would need an externally computed potential (e.g. from APBS or DelPhi) and then use the Chimera Electrostatic Surface Coloring tool (menu Tools / Surface & Binding Analysis) which uses those externally computed potential grids. Those external potential calculations can take a long time and a huge amount of memory for large models. I submitted a Chimera enhancement request to allow the Chimera built-in Coulomb coloring to work on any surface, but Eric who wrote that code has many other things to work on, so it probably won't happen soon. Tom -------- Original Message -------- Subject: SURF for large proteins From: Vijay Reddy To: Tom Goddard Date: 1/3/12 7:32 AM
Hi Tom,
Happy New Year! Wish you the best for you and yours in the New Year!
I was wondering if are there tricks to calculate and display MSMS surface for large proteins, even with the coarse representations. I am trying to display electrostatic potential.
Many thanks, Vijay
{kind=link}

A couple of notes: (A) you can color any surface by Coulombic potential with an extra step: use the Coulombic option to create a volume data set (grid of values), and then use Surface Color (or command scolor) to color the surface by that volume data. (B) other possible tricks to avoid MSMS failure are to tinker with the molecular surface calculation parameters (probe radius, vertex density) and/or slightly enlarge all atomic VDW radii. However, there is no universal recipe, it requires trial and error, not really feasible for automated work on a large number of structures. Elaine ---------- Elaine C. Meng, Ph.D. UCSF Computer Graphics Lab (Chimera team) and Babbitt Lab Department of Pharmaceutical Chemistry University of California, San Francisco On Jan 3, 2012, at 11:32 AM, Tom Goddard wrote:
Hi Vijay,
Chimera molecular surface calculation (using MSMS) fails for most molecules larger than 5,000 atoms. The main trick that can help is to split large multi-chain models into a separate model for each chain using the split command (e.g. "split #0"). Then use a separate molecular surface on each chain. The attached photo shows the result of that for chaperone 1aon with 21 chains with one subunit colored yellow so you can see its shape.
If you use Coulombic Electrostatic Coloring (menu Tools / Surface & Binding Analysis) doing it on the split model gives a slightly different result than with an unsplit model. The potential on each surface is just the potential for one chain. Doing the calculation without split would use the potential for the entire model. The split version can be more useful if you are interested in complementary charges on interfaces between chains.
Sometimes you can get a molecular surface computation that fails on one computer to work on another computer. Mac Chimera produces surfaces for larger models, and Windows Chimera has the most failures, and the failure rate for 64-bit Windows Chimera is much higher than 32-bit Windows Chimera which is higher than Mac (all versions). Not sure where Linux fits in.
Another approach is to use a different kind of surface, one generated from a density map made by placing Gaussians at each atom. The molmap command creates this surface (e.g. "molmap #0 5" for a 5 Angstrom resolution surface). You'll probably want to use a high resolution (< 5A) since the electrostatics can vary on short distances. You might want to use the molmap "gridSpacing" option to get a finer mesh for the surface (e.g. "molmap #0 5 gridSpacing 1" for a 1 Angstrom grid). Currently only molecular surfaces can be colored by Chimera's Coulomb potential coloring code, so that won't work on these molmap surfaces. You would need an externally computed potential (e.g. from APBS or DelPhi) and then use the Chimera Electrostatic Surface Coloring tool (menu Tools / Surface & Binding Analysis) which uses those externally computed potential grids. Those external potential calculations can take a long time and a huge amount of memory for large models. I submitted a Chimera enhancement request to allow the Chimera built-in Coulomb coloring to work on any surface, but Eric who wrote that code has many other things to work on, so it probably won't happen soon.
Tom
-------- Original Message -------- Subject: SURF for large proteins From: Vijay Reddy To: Tom Goddard Date: 1/3/12 7:32 AM
Hi Tom,
Happy New Year! Wish you the best for you and yours in the New Year!
I was wondering if are there tricks to calculate and display MSMS surface for large proteins, even with the coarse representations. I am trying to display electrostatic potential.
Many thanks, Vijay
<1aon-esp.jpg>_______________________________________________ Chimera-users mailing list Chimera-users@cgl.ucsf.edu http://plato.cgl.ucsf.edu/mailman/listinfo/chimera-users
participants (2)
-
 Elaine Meng
Elaine Meng -
 Tom Goddard
Tom Goddard