
Goodnight! My name is Gabrielle, I am a Master's student at UNIFESP (Federal University of São Paulo), SP, Brazil. I'm using UCSF Chimera to analyze the secondary structure of some peptides that I'm studying. When I load the .pdb file in the program, it generates a different structure than the one desired. In the examples below, we have two structures alpha helix and random coil, but the images generated are different and I don't understand why. Could you help me, explaining what is happening, or what I should do to have the image as in the first figure (NATTP_05 chimera). Thanks -- ---- A Universidade Federal de São Paulo segue a política de e-mail Institucional disponível em https://sti.unifesp.br/documentos/termos-de-uso-do-email <https://sti.unifesp.br/documentos/termos-de-uso-do-email> <https://sei.unifesp.br/sei/publicacoes/controlador_publicacoes.php?acao=publ...>
{kind=link}
{kind=link}

Hello Gabrielle, Just hide the atoms and show the ribbon. For example, menu: Actions... Atoms/Bonds... hide Actions... Ribbons... show Or, if you want to use commands instead of the menu: ~display ribbon See command help: <https://www.rbvi.ucsf.edu/chimera/docs/UsersGuide/framecommand.html> To learn Chimera, you might be interested in trying some of the tutorials: <https://www.rbvi.ucsf.edu/Outreach/Tutorials/GettingStarted.html> <https://www.rbvi.ucsf.edu/chimera/tutorials.html> If you mean that when you first open the structure it looks different, it might be because there are different numbers of atoms, and that is one of the ways Chimera tries to guess which type of display might be most useful. But if you don't want the style you get, you can just change it with the menu or commands as explained above. If you really need it to look that way right when you open it, without changing it yourself, use Preferences (menu: Favorites... Preferences), category "New Molecules" -- there you can set "smart initial display" (the guessing stuff) to "false" and "ribbon display" to "on" and "show atoms" to "false"; you would probably also want atom style "endcap" and bond style "stick" in case you decide to show some atoms later. If you want to keep these settings for the next time you use Chimera, remember to click Save on the Preferences dialog. New Molecules Preferences: <https://www.rbvi.ucsf.edu/chimera/docs/UsersGuide/preferences.html#New%20Mol...> I hope this helps, Elaine ----- Elaine C. Meng, Ph.D. UCSF Chimera(X) team Department of Pharmaceutical Chemistry University of California, San Francisco
On May 9, 2022, at 5:09 PM, GABRIELLE LUPETI DE CENA via Chimera-users <chimera-users@cgl.ucsf.edu> wrote:
Goodnight!
My name is Gabrielle, I am a Master's student at UNIFESP (Federal University of São Paulo), SP, Brazil. I'm using UCSF Chimera to analyze the secondary structure of some peptides that I'm studying.
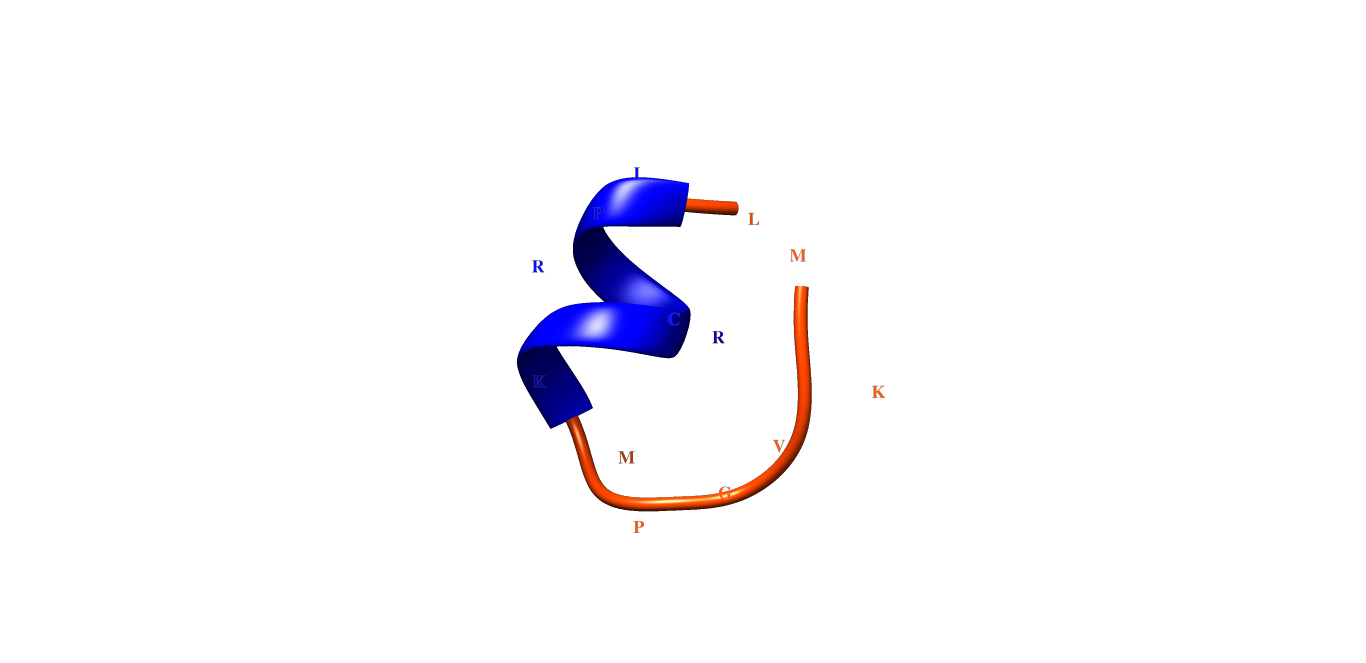
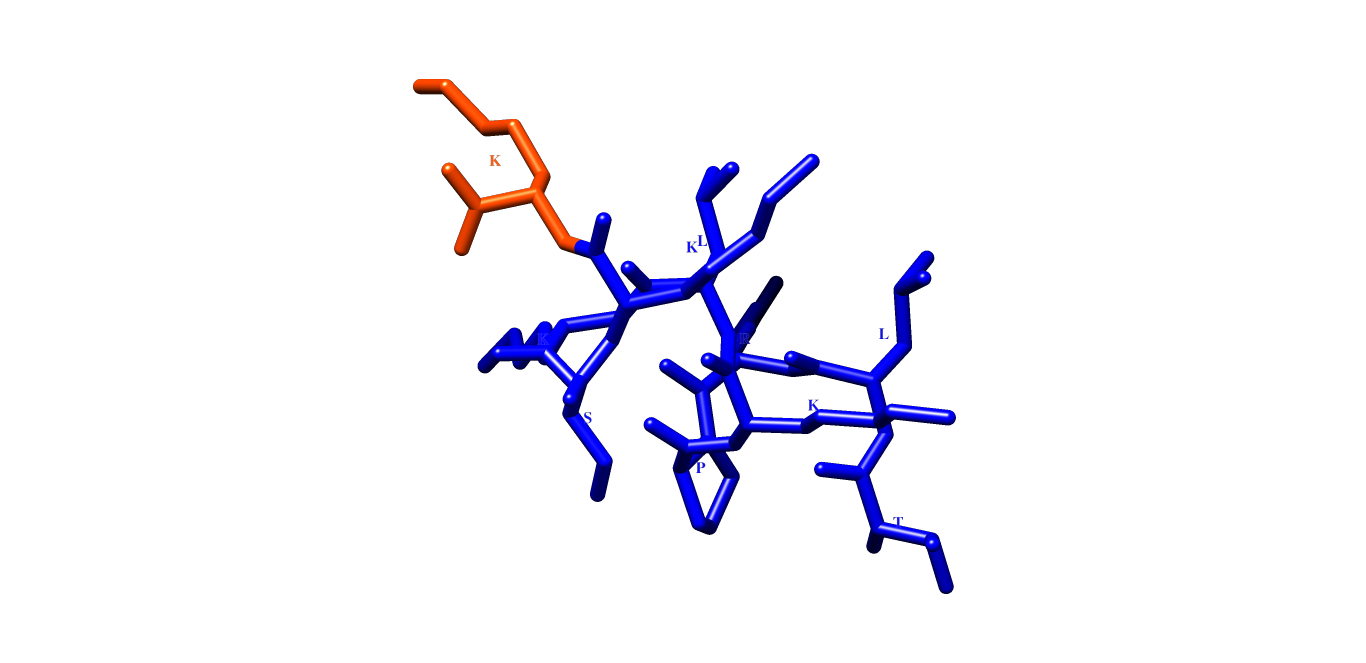
When I load the .pdb file in the program, it generates a different structure than the one desired. In the examples below, we have two structures alpha helix and random coil, but the images generated are different and I don't understand why.
Could you help me, explaining what is happening, or what I should do to have the image as in the first figure (NATTP_05 chimera). Thanks
Hello Elaine, Thank you very much for your attention! I managed to solve the problem ♥ Em seg., 9 de mai. de 2022 às 21:26, Elaine Meng <meng@cgl.ucsf.edu> escreveu:
Hello Gabrielle, Just hide the atoms and show the ribbon. For example, menu:
Actions... Atoms/Bonds... hide Actions... Ribbons... show
Or, if you want to use commands instead of the menu:
~display ribbon
See command help: <https://www.rbvi.ucsf.edu/chimera/docs/UsersGuide/framecommand.html>
To learn Chimera, you might be interested in trying some of the tutorials: <https://www.rbvi.ucsf.edu/Outreach/Tutorials/GettingStarted.html> <https://www.rbvi.ucsf.edu/chimera/tutorials.html>
If you mean that when you first open the structure it looks different, it might be because there are different numbers of atoms, and that is one of the ways Chimera tries to guess which type of display might be most useful. But if you don't want the style you get, you can just change it with the menu or commands as explained above.
If you really need it to look that way right when you open it, without changing it yourself, use Preferences (menu: Favorites... Preferences), category "New Molecules" -- there you can set "smart initial display" (the guessing stuff) to "false" and "ribbon display" to "on" and "show atoms" to "false"; you would probably also want atom style "endcap" and bond style "stick" in case you decide to show some atoms later. If you want to keep these settings for the next time you use Chimera, remember to click Save on the Preferences dialog.
New Molecules Preferences: < https://www.rbvi.ucsf.edu/chimera/docs/UsersGuide/preferences.html#New%20Mol...
I hope this helps, Elaine ----- Elaine C. Meng, Ph.D. UCSF Chimera(X) team Department of Pharmaceutical Chemistry University of California, San Francisco
On May 9, 2022, at 5:09 PM, GABRIELLE LUPETI DE CENA via Chimera-users < chimera-users@cgl.ucsf.edu> wrote:
Goodnight!
My name is Gabrielle, I am a Master's student at UNIFESP (Federal University of São Paulo), SP, Brazil. I'm using UCSF Chimera to analyze the secondary structure of some peptides that I'm studying.
When I load the .pdb file in the program, it generates a different structure than the one desired. In the examples below, we have two structures alpha helix and random coil, but the images generated are different and I don't understand why.
Could you help me, explaining what is happening, or what I should do to have the image as in the first figure (NATTP_05 chimera). Thanks
-- ---- A Universidade Federal de São Paulo segue a política de e-mail Institucional disponível em https://sti.unifesp.br/documentos/termos-de-uso-do-email <https://sti.unifesp.br/documentos/termos-de-uso-do-email> <https://sei.unifesp.br/sei/publicacoes/controlador_publicacoes.php?acao=publ...>
participants (2)
-
 Elaine Meng
Elaine Meng -
 GABRIELLE LUPETI DE CENA
GABRIELLE LUPETI DE CENA