
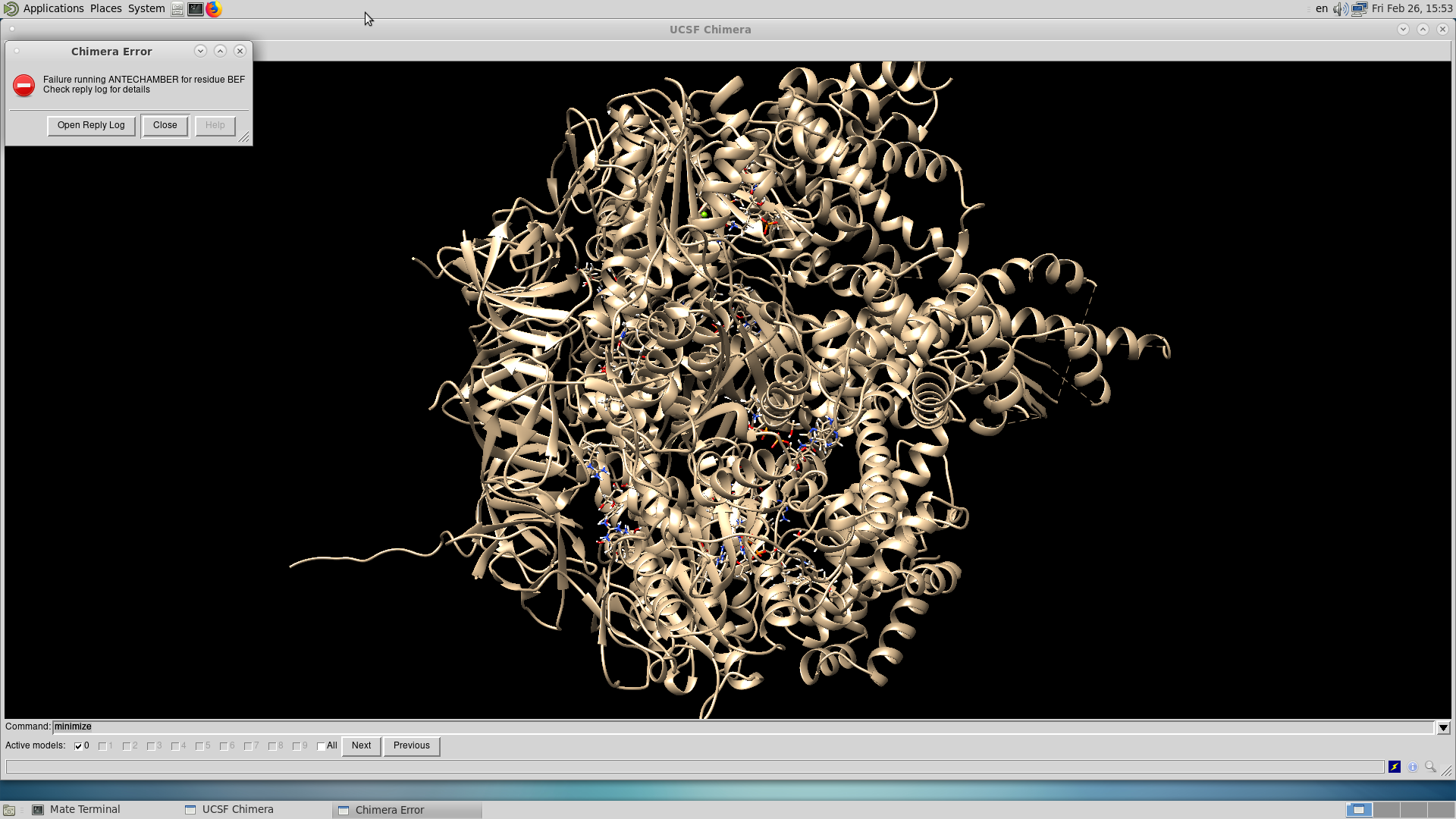
To whom it may concern: I am trying to use Chimera for protein Energy Minimization. I encountered a problem in running the program. Please find the attached screenshots of the error details. The problem seemed to be with the ligand BEF (beryllium trifluoride). Any help would be greatly appreciated. Many thanks, Mailyn
{kind=link}
{kind=link}
{kind=link}

Hello Mailyn, Probably the charge calculation step (Antechamber) cannot handle BeF3, sorry. From the Add Charge manual page "Note that Antechamber/GAFF are meant to handle most small organic molecules, but not metal complexes, inorganic compounds, or unstable species such as radicals, and may not work well on highly charged molecules. GAFF allows for parametrization of most organic molecules made of C, N, O, H, S, P, F, Cl, Br and I." As you know, BEF contains another element not in that list, Be. You may be able to minimize if you delete the BEF residue first, but I don't know if that would be useful for your project. Best, Elaine ----- Elaine C. Meng, Ph.D. UCSF Chimera(X) team Department of Pharmaceutical Chemistry University of California, San Francisco P.S. in general when you get an error message, you should not e-mail this help list but instead use Chimera menu: Help... Report a Bug, and in the bug report attach your session so that we can try your exact structure, include a short description of what you did, and also your e-mail address if you are hoping for a response
On Mar 5, 2021, at 12:18 PM, Mailyn Terrado <mailyn.terrado@rosalindfranklin.edu> wrote:
To whom it may concern:
I am trying to use Chimera for protein Energy Minimization. I encountered a problem in running the program. Please find the attached screenshots of the error details. The problem seemed to be with the ligand BEF (beryllium trifluoride).
Any help would be greatly appreciated.
Many thanks, Mailyn <Screenshot at 2021-02-26 15-53-49.png><Screenshot at 2021-02-26 15-55-21.png><Screenshot at 2021-02-26 15-55-46.png>
Hi Elaine, Thank you for your reply. I will try to remove BeF3 and re-run. I also encountered the same error with a different protein structure with the ligand ANP (phosphoaminophosphonic acid-adenyl ester, Formula: C10 H17 N 6 O12 P3. Thank you for the help. Regards, Mailyn Regards, Mailyn On Fri, Mar 5, 2021 at 2:37 PM Elaine Meng <meng@cgl.ucsf.edu> wrote:
Hello Mailyn, Probably the charge calculation step (Antechamber) cannot handle BeF3, sorry. From the Add Charge manual page
"Note that Antechamber/GAFF are meant to handle most small organic molecules, but not metal complexes, inorganic compounds, or unstable species such as radicals, and may not work well on highly charged molecules. GAFF allows for parametrization of most organic molecules made of C, N, O, H, S, P, F, Cl, Br and I."
As you know, BEF contains another element not in that list, Be. You may be able to minimize if you delete the BEF residue first, but I don't know if that would be useful for your project. Best, Elaine ----- Elaine C. Meng, Ph.D. UCSF Chimera(X) team Department of Pharmaceutical Chemistry University of California, San Francisco
P.S. in general when you get an error message, you should not e-mail this help list but instead use Chimera menu: Help... Report a Bug, and in the bug report attach your session so that we can try your exact structure, include a short description of what you did, and also your e-mail address if you are hoping for a response
On Mar 5, 2021, at 12:18 PM, Mailyn Terrado < mailyn.terrado@rosalindfranklin.edu> wrote:
To whom it may concern:
I am trying to use Chimera for protein Energy Minimization. I encountered a problem in running the program. Please find the attached screenshots of the error details. The problem seemed to be with the ligand BEF (beryllium trifluoride).
Any help would be greatly appreciated.
Many thanks, Mailyn <Screenshot at 2021-02-26 15-53-49.png><Screenshot at 2021-02-26 15-55-21.png><Screenshot at 2021-02-26 15-55-46.png>
As mentioned in the previous reply, the charge calculation may fail on highly charged structures (like phosphoesters). <https://www.rbvi.ucsf.edu/chimera/docs/ContributedSoftware/addcharge/addchar...> Elaine
On Mar 5, 2021, at 1:00 PM, Mailyn Terrado <mailyn.terrado@rosalindfranklin.edu> wrote:
Hi Elaine,
Thank you for your reply. I will try to remove BeF3 and re-run.
I also encountered the same error with a different protein structure with the ligand ANP (phosphoaminophosphonic acid-adenyl ester, Formula: C10 H17 N6 O12 P3.
Thank you for the help.
Regards, Mailyn
Regards, Mailyn
On Fri, Mar 5, 2021 at 2:37 PM Elaine Meng <meng@cgl.ucsf.edu> wrote: Hello Mailyn, Probably the charge calculation step (Antechamber) cannot handle BeF3, sorry. From the Add Charge manual page
"Note that Antechamber/GAFF are meant to handle most small organic molecules, but not metal complexes, inorganic compounds, or unstable species such as radicals, and may not work well on highly charged molecules. GAFF allows for parametrization of most organic molecules made of C, N, O, H, S, P, F, Cl, Br and I."
As you know, BEF contains another element not in that list, Be. You may be able to minimize if you delete the BEF residue first, but I don't know if that would be useful for your project. Best, Elaine ----- Elaine C. Meng, Ph.D. UCSF Chimera(X) team Department of Pharmaceutical Chemistry University of California, San Francisco
P.S. in general when you get an error message, you should not e-mail this help list but instead use Chimera menu: Help... Report a Bug, and in the bug report attach your session so that we can try your exact structure, include a short description of what you did, and also your e-mail address if you are hoping for a response
On Mar 5, 2021, at 12:18 PM, Mailyn Terrado <mailyn.terrado@rosalindfranklin.edu> wrote:
To whom it may concern:
I am trying to use Chimera for protein Energy Minimization. I encountered a problem in running the program. Please find the attached screenshots of the error details. The problem seemed to be with the ligand BEF (beryllium trifluoride).
Any help would be greatly appreciated.
Many thanks, Mailyn <Screenshot at 2021-02-26 15-53-49.png><Screenshot at 2021-02-26 15-55-21.png><Screenshot at 2021-02-26 15-55-46.png>
Chimera-users mailing list: Chimera-users@cgl.ucsf.edu Manage subscription: https://plato.cgl.ucsf.edu/mailman/listinfo/chimera-users
Hi Elaine, I removed the BEF ligand from the structure. I encountered this error message when trying to Minimize the structure. I appreciate all the help. Regards, Mailyn On Fri, Mar 5, 2021 at 3:09 PM Elaine Meng <meng@cgl.ucsf.edu> wrote:
As mentioned in the previous reply, the charge calculation may fail on highly charged structures (like phosphoesters).
< https://www.rbvi.ucsf.edu/chimera/docs/ContributedSoftware/addcharge/addchar...
Elaine
On Mar 5, 2021, at 1:00 PM, Mailyn Terrado < mailyn.terrado@rosalindfranklin.edu> wrote:
Hi Elaine,
Thank you for your reply. I will try to remove BeF3 and re-run.
I also encountered the same error with a different protein structure with the ligand ANP (phosphoaminophosphonic acid-adenyl ester, Formula: C10 H17 N6 O12 P3.
Thank you for the help.
Regards, Mailyn
Regards, Mailyn
On Fri, Mar 5, 2021 at 2:37 PM Elaine Meng <meng@cgl.ucsf.edu> wrote: Hello Mailyn, Probably the charge calculation step (Antechamber) cannot handle BeF3, sorry. From the Add Charge manual page
"Note that Antechamber/GAFF are meant to handle most small organic molecules, but not metal complexes, inorganic compounds, or unstable species such as radicals, and may not work well on highly charged molecules. GAFF allows for parametrization of most organic molecules made of C, N, O, H, S, P, F, Cl, Br and I."
As you know, BEF contains another element not in that list, Be. You may be able to minimize if you delete the BEF residue first, but I don't know if that would be useful for your project. Best, Elaine ----- Elaine C. Meng, Ph.D. UCSF Chimera(X) team Department of Pharmaceutical Chemistry University of California, San Francisco
P.S. in general when you get an error message, you should not e-mail this help list but instead use Chimera menu: Help... Report a Bug, and in the bug report attach your session so that we can try your exact structure, include a short description of what you did, and also your e-mail address if you are hoping for a response
On Mar 5, 2021, at 12:18 PM, Mailyn Terrado < mailyn.terrado@rosalindfranklin.edu> wrote:
To whom it may concern:
I am trying to use Chimera for protein Energy Minimization. I encountered a problem in running the program. Please find the attached screenshots of the error details. The problem seemed to be with the ligand BEF (beryllium trifluoride).
Any help would be greatly appreciated.
Many thanks, Mailyn <Screenshot at 2021-02-26 15-53-49.png><Screenshot at 2021-02-26 15-55-21.png><Screenshot at 2021-02-26 15-55-46.png>
Chimera-users mailing list: Chimera-users@cgl.ucsf.edu Manage subscription: https://plato.cgl.ucsf.edu/mailman/listinfo/chimera-users
{kind=link}
{kind=link}
Hi Mailyn, Error tracebacks should not be reported to this e-mail address. Instead please use menu: Help... Report a Bug in the menu, and attach your a Chimera session file to the report so that we can try your exact structure. However, it may be that the structure still contains some molecules that simply cannot be handled by Antechamber, and we would not be able to fix that, sorry. The link I sent before describes that issue:.
On Fri, Mar 5, 2021 at 3:09 PM Elaine Meng <meng@cgl.ucsf.edu> wrote: As mentioned in the previous reply, the charge calculation may fail on highly charged structures (like phosphoesters).
<https://www.rbvi.ucsf.edu/chimera/docs/ContributedSoftware/addcharge/addchar...>
Thanks Elaine ----- Elaine C. Meng, Ph.D. UCSF Chimera(X) team Department of Pharmaceutical Chemistry University of California, San Francisco
On Mar 12, 2021, at 11:20 AM, Mailyn Terrado <mailyn.terrado@rosalindfranklin.edu> wrote:
Hi Elaine,
I removed the BEF ligand from the structure. I encountered this error message when trying to Minimize the structure.
I appreciate all the help.
Regards, Mailyn
On Fri, Mar 5, 2021 at 3:09 PM Elaine Meng <meng@cgl.ucsf.edu> wrote: As mentioned in the previous reply, the charge calculation may fail on highly charged structures (like phosphoesters).
<https://www.rbvi.ucsf.edu/chimera/docs/ContributedSoftware/addcharge/addchar...>
Elaine
On Mar 5, 2021, at 1:00 PM, Mailyn Terrado <mailyn.terrado@rosalindfranklin.edu> wrote:
Hi Elaine,
Thank you for your reply. I will try to remove BeF3 and re-run.
I also encountered the same error with a different protein structure with the ligand ANP (phosphoaminophosphonic acid-adenyl ester, Formula: C10 H17 N6 O12 P3.
Thank you for the help.
Regards, Mailyn
Regards, Mailyn
On Fri, Mar 5, 2021 at 2:37 PM Elaine Meng <meng@cgl.ucsf.edu> wrote: Hello Mailyn, Probably the charge calculation step (Antechamber) cannot handle BeF3, sorry. From the Add Charge manual page
"Note that Antechamber/GAFF are meant to handle most small organic molecules, but not metal complexes, inorganic compounds, or unstable species such as radicals, and may not work well on highly charged molecules. GAFF allows for parametrization of most organic molecules made of C, N, O, H, S, P, F, Cl, Br and I."
As you know, BEF contains another element not in that list, Be. You may be able to minimize if you delete the BEF residue first, but I don't know if that would be useful for your project. Best, Elaine ----- Elaine C. Meng, Ph.D. UCSF Chimera(X) team Department of Pharmaceutical Chemistry University of California, San Francisco
P.S. in general when you get an error message, you should not e-mail this help list but instead use Chimera menu: Help... Report a Bug, and in the bug report attach your session so that we can try your exact structure, include a short description of what you did, and also your e-mail address if you are hoping for a response
On Mar 5, 2021, at 12:18 PM, Mailyn Terrado <mailyn.terrado@rosalindfranklin.edu> wrote:
To whom it may concern:
I am trying to use Chimera for protein Energy Minimization. I encountered a problem in running the program. Please find the attached screenshots of the error details. The problem seemed to be with the ligand BEF (beryllium trifluoride).
Any help would be greatly appreciated.
Many thanks, Mailyn <Screenshot at 2021-02-26 15-53-49.png><Screenshot at 2021-02-26 15-55-21.png><Screenshot at 2021-02-26 15-55-46.png>
participants (2)
-
 Elaine Meng
Elaine Meng -
 Mailyn Terrado
Mailyn Terrado