segmentation and resolution

To whom it may concern, I am currently working on segmenting a high resolution cryo-EM map (~4Å by the ³gold standard FSC²) to phase crystallography data. After the segmentation, my colleague doing the phasing tells me that map appears more like 12Å resolution and I am worried I am doing something wrong during the process and somehow distorting my original data. I believe the 4Å of the original map based on structural features that I can clearly see. And the segmented density does not appear as defined. Therefore it appears that there has been some changes. In order to begin the process, I did some manipulations before I started. This is an icosahedral virus map created through Auto3dem. I am only using an small quadrant of the original map, and since it was in pif format originally, I used proc3d to convert to mrc and invert the contrast to be compatible with segger. Before segmentation the map visually looked the same as the original pif map when both were displayed in chimera. The procedure I followed after segmentation was: "Save selected regions to .mrc file... save density map masked by the selected <https://www.cgl.ucsf.edu/chimera/docs/UsersGuide/selection.html> segmentation regions to an MRC file (map dimensions set to the minimal box containing the regions)². I have made sure that the correct binning has been applied during segmentation. Any help would be much appreciated. Kristin -- Kristin N. Parent, Ph.D. Assistant Professor Dept. of Biochemistry and Molecular Biology Michigan State University 603 Wilson Road Room 519 Biochemistry Building East Lansing, MI 48824 Office phone: (517) 432-8434 https://kparentlab.natsci.msu.edu/

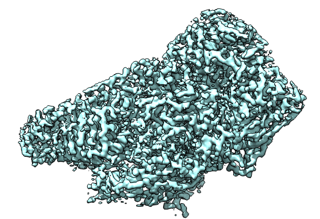
Hi Kristin, Here is my guess about why your segmented map looks lower resolution. When the Chimera Segment Map (Segger) tool writes a selected region to an MRC file it simply sets to zero all the map values outside the region surface. This creates a jagged boundary in the resulting saved map that looks horrible when you view the surface of the saved map. It will be especially bad in your case because the segmentation region surface has hundreds of tiny protrusions. A better approach is to first smooth the map, say Gaussian filter with standard deviation 5 Angstroms, segment that smooth map, giving smooth regions without all the little protrusions, then save the desired region to an MRC using the original full resolution map. This makes jagged boundary only on the outside smooth surface of the region instead of around hundreds of little protrusions. There is a trick to this procedure. To extract the full resolution density using the smoothed map regions, choose the high resolution map in the Segment Map dialog and use its menu entry File / Associate Selected to associate the smoothed map segmentation with the full resolution map. I’ve attached images showing the problem and suggested approach applied to beta-galactosidase EMDB 2984 at about 3A resolution. You still have sharp drops in the density to 0 and you may want to roll those off using the “vop falloff” command described here http://www.cgl.ucsf.edu/chimera/data/falloff-sep2012/falloff.html <http://www.cgl.ucsf.edu/chimera/data/falloff-sep2012/falloff.html> Tom 3A beta-galactosidase map Segmentation with Segment Map / Segger is too detailed. Smoothed map with Gaussian filter, 5A standard deviation: Segmentation surfaces using smoothed map Region extracted from full resolution map using segmentation from smoothed map
On Sep 23, 2016, at 7:52 AM, Kristin Parent wrote:
To whom it may concern,
I am currently working on segmenting a high resolution cryo-EM map (~4Å by the “gold standard FSC”) to phase crystallography data. After the segmentation, my colleague doing the phasing tells me that map appears more like 12Å resolution and I am worried I am doing something wrong during the process and somehow distorting my original data. I believe the 4Å of the original map based on structural features that I can clearly see. And the segmented density does not appear as defined. Therefore it appears that there has been some changes.
In order to begin the process, I did some manipulations before I started. This is an icosahedral virus map created through Auto3dem. I am only using an small quadrant of the original map, and since it was in pif format originally, I used proc3d to convert to mrc and invert the contrast to be compatible with segger. Before segmentation the map visually looked the same as the original pif map when both were displayed in chimera.
The procedure I followed after segmentation was: "Save selected regions to .mrc file... save density map masked by the selected <https://www.cgl.ucsf.edu/chimera/docs/UsersGuide/selection.html> segmentation regions to an MRC file (map dimensions set to the minimal box containing the regions)”. I have made sure that the correct binning has been applied during segmentation.
Any help would be much appreciated. Kristin
-- Kristin N. Parent, Ph.D. Assistant Professor Dept. of Biochemistry and Molecular Biology Michigan State University 603 Wilson Road Room 519 Biochemistry Building East Lansing, MI 48824 Office phone: (517) 432-8434 https://kparentlab.natsci.msu.edu <https://kparentlab.natsci.msu.edu/>/
_______________________________________________ Chimera-users mailing list: Chimera-users@cgl.ucsf.edu Manage subscription: http://plato.cgl.ucsf.edu/mailman/listinfo/chimera-users
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
participants (2)
-
 Kristin Parent
Kristin Parent -
 Tom Goddard
Tom Goddard