
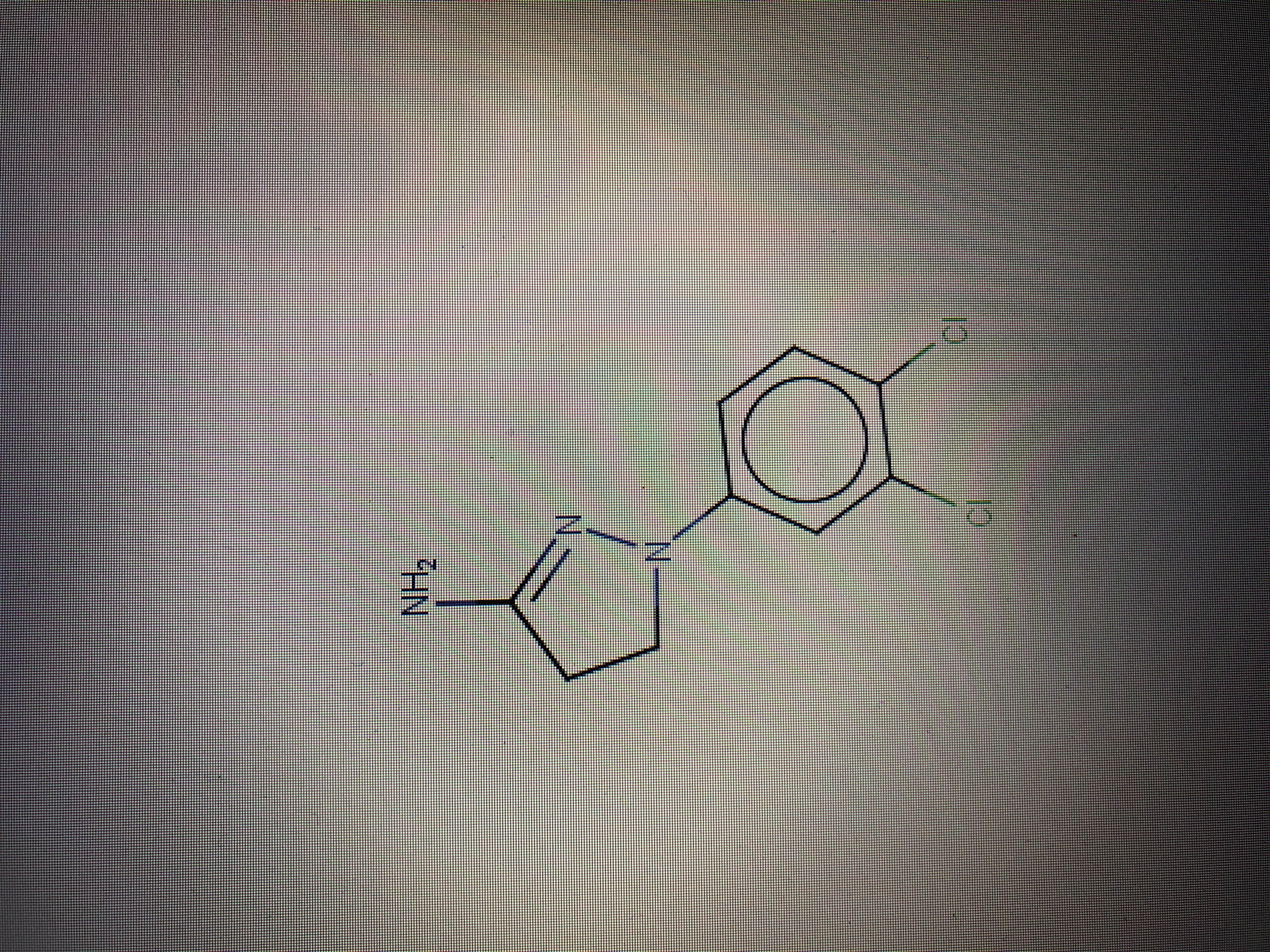
Hi, I have a problem with the result of autodock Vina. When I load a molecule in “mol2” format and I use AutoDock Vina for docking, the docked molecule is altered. How can be possible? How can I solve my problem? I attach the photo of the real molecule [cid:C2748141-718F-40C4-8964-A212C52FDB1A] And this I how the molecule changes after the docking [cid:C1EDE285-2873-437D-A034-5BB2D031BD67] The 5-term ring becomes aromatic, while it should have only a double bond. ( this happens both if I save autodock vina result in “mol2” format and “pdb” format Hope you can help me!
{kind=link}
{kind=link}

Hi Greta, I guess the only difference is whether the two carbons in the 5-member ring are CH or CH2, because Chimera does not use (or change) bond orders. Are you sure that both structures can exist as real chemicals? Maybe Chimera guesses it is an aromatic ring (more on this below) but that would also be true as soon as you opened the ligand structure, not as a result of the docking. So my question is then, how can you tell the molecule is different before and after? It would have been much more useful to attach the before and after structures (coordinate files) than images of 2D chemical diagrams. The following assumes that both structures you drew can exist as real chemicals. Did it have hydrogens before you started the docking calculation? If not, it may be that the autodock ligand preparation script added hydrogens and had trouble identifying the bond orders (maybe the bond length is similar with single or aromatic bond) and added 1 H instead of 2 to each of those atoms. In that case you could try adding hydrogens to the ligand with Chimera instead (e.g. command “addh” or AddH tool) <http://www.rbvi.ucsf.edu/chimera/docs/ContributedSoftware/addh/addh.html> before docking. If Chimera has the same problem as the autodock ligand preparation script, then delete the ligand hydrogens and make sure the atom types are assigned to sp3 carbon before trying to add them back again. For example, select those two carbon atoms (Ctrl-click, Shift-Ctrl-click) and then use setattr command, something like: setattr a idatmType C3 sel <http://www.rbvi.ucsf.edu/chimera/docs/UsersGuide/midas/setattr.html> <http://www.rbvi.ucsf.edu/chimera/docs/UsersGuide/idatm.html> Then add hydrogens, check that they are as expected, then do the docking. Or you could use any other program/method to add hydrogens before opening the ligand structure in Chimera. It may not make much difference since if I remember correctly, Vina doesn’t even use the hydrogens except to identify the types of the other atoms. You would have to check the literature references to be sure about that, however. Finally: Note that the Autodock Vina tool uses a web server that only allows very little sampling, which is not adequate for most research purposes, as mentioned in the boxed warning on the manual page. <http://www.rbvi.ucsf.edu/chimera/docs/ContributedSoftware/vina/vina.html> Best, Elaine ----- Elaine C. Meng, Ph.D. UCSF Chimera(X) team Department of Pharmaceutical Chemistry University of California, San Francisco
On Feb 7, 2020, at 2:45 PM, Greta Hodo <gretahodo96@hotmail.com> wrote:
Hi, I have a problem with the result of autodock Vina. When I load a molecule in “mol2” format and I use AutoDock Vina for docking, the docked molecule is altered. How can be possible? How can I solve my problem?
I attach the photo of the real molecule <image0.jpeg>
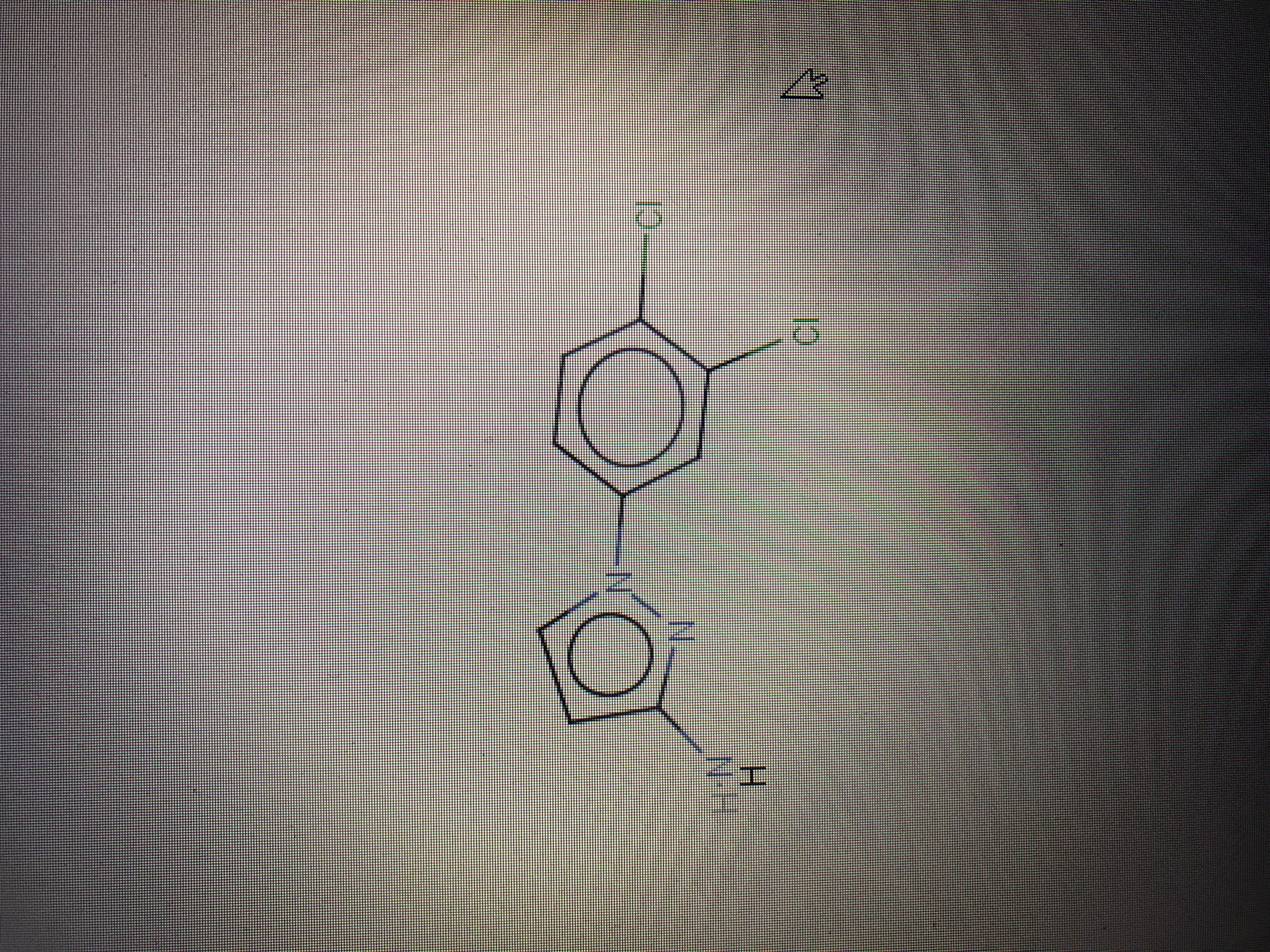
And this I how the molecule changes after the docking <image1.jpeg> The 5-term ring becomes aromatic, while it should have only a double bond. ( this happens both if I save autodock vina result in “mol2” format and “pdb” format
Hope you can help me!
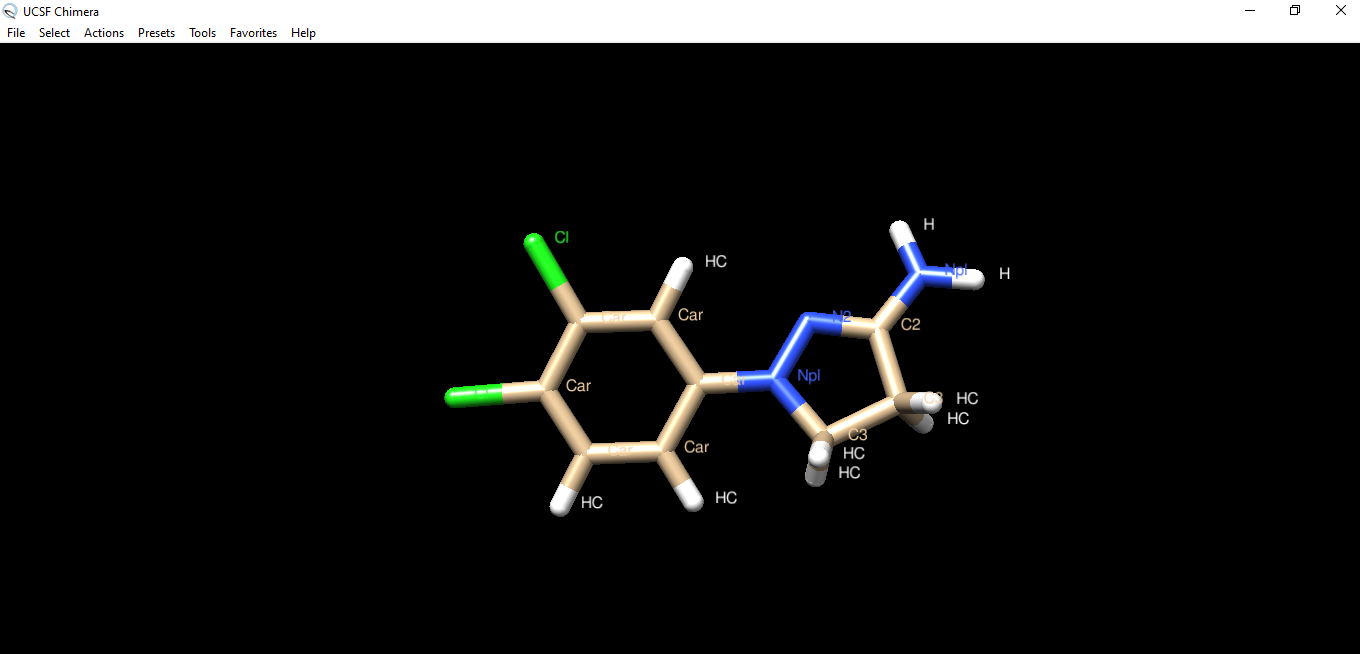
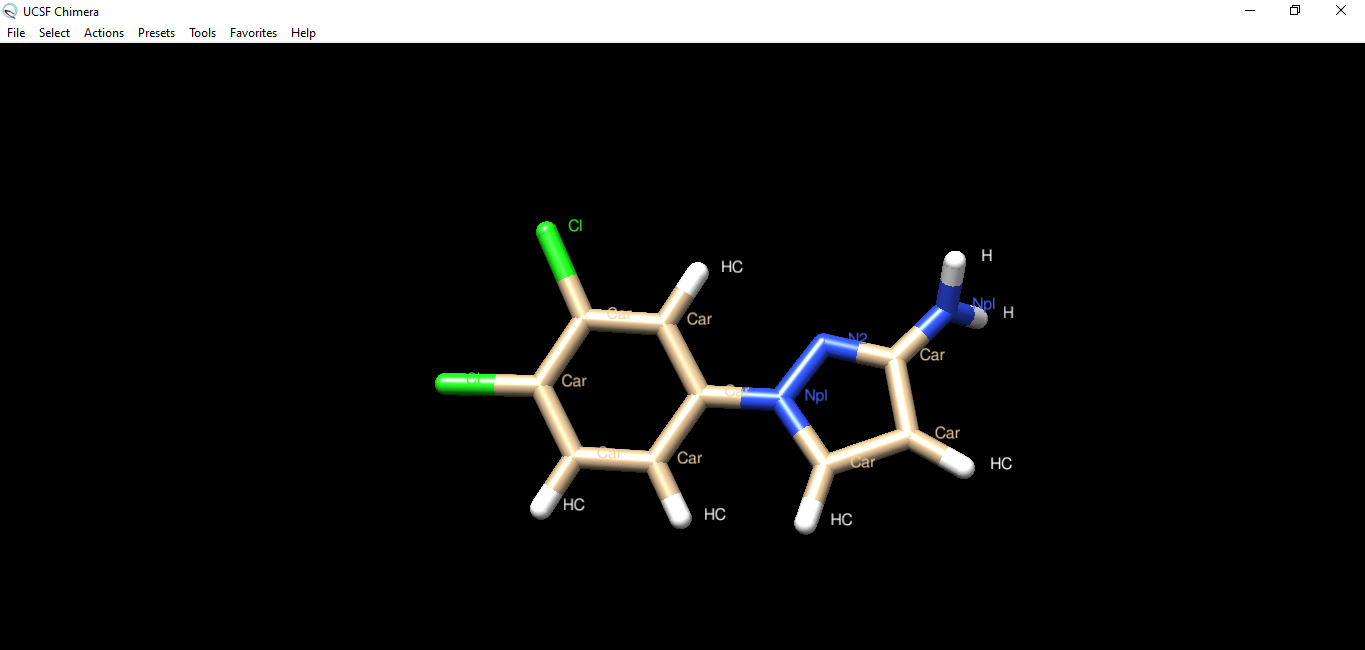
Hi Elaine, thank you for the quick answer. Before I start the docking calculation my molecule has all the hydrogens as expected (as you can see in the ''img1'' file attached you). After the docking calculation with Autodock Vina the molecule changes (as you can see in the ''img2'' file). How can I solve the problem? Best, Greta ________________________________ Da: Elaine Meng <meng@cgl.ucsf.edu> Inviato: sabato 8 febbraio 2020 01:16 A: Greta Hodo <gretahodo96@hotmail.com> Cc: chimera-users@cgl.ucsf.edu <chimera-users@cgl.ucsf.edu> Oggetto: Re: [Chimera-users] Docking Hi Greta, I guess the only difference is whether the two carbons in the 5-member ring are CH or CH2, because Chimera does not use (or change) bond orders. Are you sure that both structures can exist as real chemicals? Maybe Chimera guesses it is an aromatic ring (more on this below) but that would also be true as soon as you opened the ligand structure, not as a result of the docking. So my question is then, how can you tell the molecule is different before and after? It would have been much more useful to attach the before and after structures (coordinate files) than images of 2D chemical diagrams. The following assumes that both structures you drew can exist as real chemicals. Did it have hydrogens before you started the docking calculation? If not, it may be that the autodock ligand preparation script added hydrogens and had trouble identifying the bond orders (maybe the bond length is similar with single or aromatic bond) and added 1 H instead of 2 to each of those atoms. In that case you could try adding hydrogens to the ligand with Chimera instead (e.g. command “addh” or AddH tool) <http://www.rbvi.ucsf.edu/chimera/docs/ContributedSoftware/addh/addh.html> before docking. If Chimera has the same problem as the autodock ligand preparation script, then delete the ligand hydrogens and make sure the atom types are assigned to sp3 carbon before trying to add them back again. For example, select those two carbon atoms (Ctrl-click, Shift-Ctrl-click) and then use setattr command, something like: setattr a idatmType C3 sel <http://www.rbvi.ucsf.edu/chimera/docs/UsersGuide/midas/setattr.html> <http://www.rbvi.ucsf.edu/chimera/docs/UsersGuide/idatm.html> Then add hydrogens, check that they are as expected, then do the docking. Or you could use any other program/method to add hydrogens before opening the ligand structure in Chimera. It may not make much difference since if I remember correctly, Vina doesn’t even use the hydrogens except to identify the types of the other atoms. You would have to check the literature references to be sure about that, however. Finally: Note that the Autodock Vina tool uses a web server that only allows very little sampling, which is not adequate for most research purposes, as mentioned in the boxed warning on the manual page. <http://www.rbvi.ucsf.edu/chimera/docs/ContributedSoftware/vina/vina.html> Best, Elaine ----- Elaine C. Meng, Ph.D. UCSF Chimera(X) team Department of Pharmaceutical Chemistry University of California, San Francisco
On Feb 7, 2020, at 2:45 PM, Greta Hodo <gretahodo96@hotmail.com> wrote:
Hi, I have a problem with the result of autodock Vina. When I load a molecule in “mol2” format and I use AutoDock Vina for docking, the docked molecule is altered. How can be possible? How can I solve my problem?
I attach the photo of the real molecule <image0.jpeg>
And this I how the molecule changes after the docking <image1.jpeg> The 5-term ring becomes aromatic, while it should have only a double bond. ( this happens both if I save autodock vina result in “mol2” format and “pdb” format
Hope you can help me!
{kind=link}
{kind=link}
Hi Greta, In the “Ligand options” try turning off “Merge charges and remove non-polar hydrogens,” i.e. set it to “false”: <http://www.rbvi.ucsf.edu/chimera/docs/ContributedSoftware/vina/vina.html> That’s my only idea at this point. However, it still doesn’t make sense because if you had it set to “true” there should not be any hydrogens on the carbons in the output. Elaine
On Feb 7, 2020, at 5:11 PM, Greta Hodo <gretahodo96@hotmail.com> wrote:
Hi Elaine, thank you for the quick answer. Before I start the docking calculation my molecule has all the hydrogens as expected (as you can see in the ''img1'' file attached you). After the docking calculation with Autodock Vina the molecule changes (as you can see in the ''img2'' file). How can I solve the problem?
Best, Greta
Da: Elaine Meng <meng@cgl.ucsf.edu> Inviato: sabato 8 febbraio 2020 01:16 A: Greta Hodo <gretahodo96@hotmail.com> Cc: chimera-users@cgl.ucsf.edu <chimera-users@cgl.ucsf.edu> Oggetto: Re: [Chimera-users] Docking
Hi Greta, I guess the only difference is whether the two carbons in the 5-member ring are CH or CH2, because Chimera does not use (or change) bond orders. Are you sure that both structures can exist as real chemicals? Maybe Chimera guesses it is an aromatic ring (more on this below) but that would also be true as soon as you opened the ligand structure, not as a result of the docking. So my question is then, how can you tell the molecule is different before and after? It would have been much more useful to attach the before and after structures (coordinate files) than images of 2D chemical diagrams.
The following assumes that both structures you drew can exist as real chemicals.
Did it have hydrogens before you started the docking calculation? If not, it may be that the autodock ligand preparation script added hydrogens and had trouble identifying the bond orders (maybe the bond length is similar with single or aromatic bond) and added 1 H instead of 2 to each of those atoms. In that case you could try adding hydrogens to the ligand with Chimera instead (e.g. command “addh” or AddH tool) <http://www.rbvi.ucsf.edu/chimera/docs/ContributedSoftware/addh/addh.html> before docking. If Chimera has the same problem as the autodock ligand preparation script, then delete the ligand hydrogens and make sure the atom types are assigned to sp3 carbon before trying to add them back again. For example, select those two carbon atoms (Ctrl-click, Shift-Ctrl-click) and then use setattr command, something like:
setattr a idatmType C3 sel
<http://www.rbvi.ucsf.edu/chimera/docs/UsersGuide/midas/setattr.html> <http://www.rbvi.ucsf.edu/chimera/docs/UsersGuide/idatm.html>
Then add hydrogens, check that they are as expected, then do the docking. Or you could use any other program/method to add hydrogens before opening the ligand structure in Chimera.
It may not make much difference since if I remember correctly, Vina doesn’t even use the hydrogens except to identify the types of the other atoms. You would have to check the literature references to be sure about that, however.
Finally: Note that the Autodock Vina tool uses a web server that only allows very little sampling, which is not adequate for most research purposes, as mentioned in the boxed warning on the manual page. <http://www.rbvi.ucsf.edu/chimera/docs/ContributedSoftware/vina/vina.html>
Best, Elaine ----- Elaine C. Meng, Ph.D. UCSF Chimera(X) team Department of Pharmaceutical Chemistry University of California, San Francisco
On Feb 7, 2020, at 2:45 PM, Greta Hodo <gretahodo96@hotmail.com> wrote:
Hi, I have a problem with the result of autodock Vina. When I load a molecule in “mol2” format and I use AutoDock Vina for docking, the docked molecule is altered. How can be possible? How can I solve my problem?
I attach the photo of the real molecule <image0.jpeg>
And this I how the molecule changes after the docking <image1.jpeg> The 5-term ring becomes aromatic, while it should have only a double bond. ( this happens both if I save autodock vina result in “mol2” format and “pdb” format
Hope you can help me!
<img1.png><img2.png>_______________________________________________ Chimera-users mailing list: Chimera-users@cgl.ucsf.edu Manage subscription: http://plato.cgl.ucsf.edu/mailman/listinfo/chimera-users
Hi Elaine, I did as you suggested me and I solved the problem, so thank you very much! Thank you for your time and for your quick answers Best, Greta
Il giorno 10 feb 2020, alle ore 19:17, Elaine Meng <meng@cgl.ucsf.edu> ha scritto:
Hi Greta, In the “Ligand options” try turning off “Merge charges and remove non-polar hydrogens,” i.e. set it to “false”:
<http://www.rbvi.ucsf.edu/chimera/docs/ContributedSoftware/vina/vina.html>
That’s my only idea at this point. However, it still doesn’t make sense because if you had it set to “true” there should not be any hydrogens on the carbons in the output.
Elaine
On Feb 7, 2020, at 5:11 PM, Greta Hodo <gretahodo96@hotmail.com> wrote:
Hi Elaine, thank you for the quick answer. Before I start the docking calculation my molecule has all the hydrogens as expected (as you can see in the ''img1'' file attached you). After the docking calculation with Autodock Vina the molecule changes (as you can see in the ''img2'' file). How can I solve the problem?
Best, Greta
Da: Elaine Meng <meng@cgl.ucsf.edu> Inviato: sabato 8 febbraio 2020 01:16 A: Greta Hodo <gretahodo96@hotmail.com> Cc: chimera-users@cgl.ucsf.edu <chimera-users@cgl.ucsf.edu> Oggetto: Re: [Chimera-users] Docking
Hi Greta, I guess the only difference is whether the two carbons in the 5-member ring are CH or CH2, because Chimera does not use (or change) bond orders. Are you sure that both structures can exist as real chemicals? Maybe Chimera guesses it is an aromatic ring (more on this below) but that would also be true as soon as you opened the ligand structure, not as a result of the docking. So my question is then, how can you tell the molecule is different before and after? It would have been much more useful to attach the before and after structures (coordinate files) than images of 2D chemical diagrams.
The following assumes that both structures you drew can exist as real chemicals.
Did it have hydrogens before you started the docking calculation? If not, it may be that the autodock ligand preparation script added hydrogens and had trouble identifying the bond orders (maybe the bond length is similar with single or aromatic bond) and added 1 H instead of 2 to each of those atoms. In that case you could try adding hydrogens to the ligand with Chimera instead (e.g. command “addh” or AddH tool) <http://www.rbvi.ucsf.edu/chimera/docs/ContributedSoftware/addh/addh.html> before docking. If Chimera has the same problem as the autodock ligand preparation script, then delete the ligand hydrogens and make sure the atom types are assigned to sp3 carbon before trying to add them back again. For example, select those two carbon atoms (Ctrl-click, Shift-Ctrl-click) and then use setattr command, something like:
setattr a idatmType C3 sel
<http://www.rbvi.ucsf.edu/chimera/docs/UsersGuide/midas/setattr.html> <http://www.rbvi.ucsf.edu/chimera/docs/UsersGuide/idatm.html>
Then add hydrogens, check that they are as expected, then do the docking. Or you could use any other program/method to add hydrogens before opening the ligand structure in Chimera.
It may not make much difference since if I remember correctly, Vina doesn’t even use the hydrogens except to identify the types of the other atoms. You would have to check the literature references to be sure about that, however.
Finally: Note that the Autodock Vina tool uses a web server that only allows very little sampling, which is not adequate for most research purposes, as mentioned in the boxed warning on the manual page. <http://www.rbvi.ucsf.edu/chimera/docs/ContributedSoftware/vina/vina.html>
Best, Elaine ----- Elaine C. Meng, Ph.D. UCSF Chimera(X) team Department of Pharmaceutical Chemistry University of California, San Francisco
On Feb 7, 2020, at 2:45 PM, Greta Hodo <gretahodo96@hotmail.com> wrote:
Hi, I have a problem with the result of autodock Vina. When I load a molecule in “mol2” format and I use AutoDock Vina for docking, the docked molecule is altered. How can be possible? How can I solve my problem?
I attach the photo of the real molecule <image0.jpeg>
And this I how the molecule changes after the docking <image1.jpeg> The 5-term ring becomes aromatic, while it should have only a double bond. ( this happens both if I save autodock vina result in “mol2” format and “pdb” format
Hope you can help me!
<img1.png><img2.png>_______________________________________________ Chimera-users mailing list: Chimera-users@cgl.ucsf.edu Manage subscription: http://plato.cgl.ucsf.edu/mailman/listinfo/chimera-users
participants (2)
-
 Elaine Meng
Elaine Meng -
 Greta Hodo
Greta Hodo