
Hello...for my Master's dissertation, I need to substitute a sulphur atom for an oxygen in a Molecular Dynamics simulation. The run works fine with my target molecule 1KF1.pdb uploaded into Chimera, but the substituted version sends quite a few error messages and won't run. It doesn't appear to recognise the novel S atom. Any help would be much appreciated...I've been stuck on this for nearly a week! Best wishes Simon

Hi Simon, It would have been helpful if you said how you substituted S for O and what the error messages were. I have no idea what you did. You can change atom type and element inside of Chimera or use a text editor (not in Chimera) to change the PDB file before opening it in Chimera. The way to change the atom inside of Chimera is to select the atom and then use Build Structure (in menu under Tools… Structure Editing), the Modify Structure section. Use the option to change the name of the residue since if you keep the name the same, Chimera is expecting that residue, not something different with a sulphur in it. <http://www.rbvi.ucsf.edu/chimera/docs/ContributedSoftware/editing/editing.ht...> If you try text-editing instead, remember spacing is important in PDB files, so don’t change the spacing. There's a summary of PDB format here: <http://www.rbvi.ucsf.edu/chimera/docs/UsersGuide/tutorials/framepdbintro.htm...> In the text-editor of your choice, in the ATOM line for that atom, I would change the atom name, the element symbol (if present, would be near the end of the line), and in the ATOM lines for the whole residue, change the residue name. Regardless of how you change the atom, however, another problem is that this nonstandard residue must be parametrized if you are going to run MD. Chimera will try to do this automatically using AMBER’s Antechamber module, but especially with highly charged residues such as nucleotides it may fail. In that case, I don’t really have any solution other than to try the simpler Gasteiger charges if it gives you a choice of Gasteiger or AM1-BCC. <http://www.rbvi.ucsf.edu/chimera/docs/ContributedSoftware/addcharge/addcharg...> I hope this helps, Elaine ----- Elaine C. Meng, Ph.D. UCSF Chimera(X) team Department of Pharmaceutical Chemistry University of California, San Francisco
On Dec 27, 2017, at 1:49 PM, simon chapman <rowanlodge19@gmail.com> wrote:
Hello...for my Master's dissertation, I need to substitute a sulphur atom for an oxygen in a Molecular Dynamics simulation. The run works fine with my target molecule 1KF1.pdb uploaded into Chimera, but the substituted version sends quite a few error messages and won't run. It doesn't appear to recognise the novel S atom.
Any help would be much appreciated...I've been stuck on this for nearly a week!
Best wishes Simon
Hello Elaine. Yes,remiss of me to leave out so much info! I'm new-ish to Chimera and wasn't really sure if there would be a reply (as has happened with some other MD products) So thanks for your prompt response. I had no problems with Build Structure after being pointed in the right direction. I will try text editor eventually just for the experience. I have successfully substituted S for O in a guanine complex, and will extend that to a Se substitution later. The MD simulation runs *much* faster if I exclude the Periodic Boundary Conditions option. Is there a way to extend the run-time? Currently it covers 1000fs. Is getting to picosecond region a possibility? Best wishes and congrats to whoever put Chimera MD together! Simon On 28 December 2017 at 17:09, Elaine Meng <meng@cgl.ucsf.edu> wrote:
Hi Simon, It would have been helpful if you said how you substituted S for O and what the error messages were. I have no idea what you did.
You can change atom type and element inside of Chimera or use a text editor (not in Chimera) to change the PDB file before opening it in Chimera.
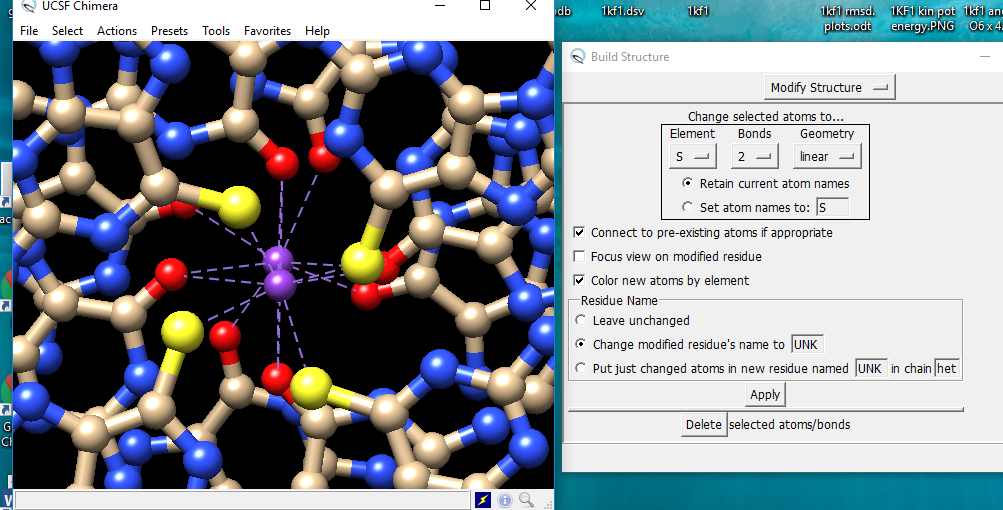
The way to change the atom inside of Chimera is to select the atom and then use Build Structure (in menu under Tools… Structure Editing), the Modify Structure section. Use the option to change the name of the residue since if you keep the name the same, Chimera is expecting that residue, not something different with a sulphur in it. <http://www.rbvi.ucsf.edu/chimera/docs/ContributedSoftware/editing/ editing.html#modify>
If you try text-editing instead, remember spacing is important in PDB files, so don’t change the spacing. There's a summary of PDB format here: <http://www.rbvi.ucsf.edu/chimera/docs/UsersGuide/ tutorials/framepdbintro.html>
In the text-editor of your choice, in the ATOM line for that atom, I would change the atom name, the element symbol (if present, would be near the end of the line), and in the ATOM lines for the whole residue, change the residue name.
Regardless of how you change the atom, however, another problem is that this nonstandard residue must be parametrized if you are going to run MD. Chimera will try to do this automatically using AMBER’s Antechamber module, but especially with highly charged residues such as nucleotides it may fail. In that case, I don’t really have any solution other than to try the simpler Gasteiger charges if it gives you a choice of Gasteiger or AM1-BCC. <http://www.rbvi.ucsf.edu/chimera/docs/ContributedSoftware/addcharge/ addcharge.html#antechamber>
I hope this helps, Elaine ----- Elaine C. Meng, Ph.D. UCSF Chimera(X) team Department of Pharmaceutical Chemistry University of California, San Francisco
On Dec 27, 2017, at 1:49 PM, simon chapman <rowanlodge19@gmail.com> wrote:
Hello...for my Master's dissertation, I need to substitute a sulphur atom for an oxygen in a Molecular Dynamics simulation. The run works fine with my target molecule 1KF1.pdb uploaded into Chimera, but the substituted version sends quite a few error messages and won't run. It doesn't appear to recognise the novel S atom.
Any help would be much appreciated...I've been stuck on this for nearly a week!
Best wishes Simon
Hi Simon, The calculation runs on your own computer and as far as I know, there isn’t a limit on the run time. The “Run Parameters” section of the Molecular Dynamics SImulation dialog allows you to enter the numbers of steps for minimization, equilibration, and production. If you click on “minimization” you can see it is actually a little a menu that can switch to “equilibration” and “production” subsections. <http://www.rbvi.ucsf.edu/chimera/docs/ContributedSoftware/md/md.html#run> You can also specify a restart file for the production run, for subsequently doing another production run starting from the endpoint of the current one. As far as I know, there isn’t an upper limit to the number of steps per run, but it might be wiser to do multiple production runs anyway (so that if something happens, you don’t lose so much prior computation). The tool was intended more to make MD simulations accessible to many people without too steep of a learning curve, rather than for very large-scale, long simulations which might be accomplished more efficiently with a dedicated MD package (AMBER, GROMACS, etc.), but you can certainly try. I hope this helps, Elaine ----- Elaine C. Meng, Ph.D. UCSF Chimera(X) team Department of Pharmaceutical Chemistry University of California, San Francisco
On Dec 30, 2017, at 4:10 AM, simon chapman <rowanlodge19@gmail.com> wrote:
Hello Elaine. Yes,remiss of me to leave out so much info! I'm new-ish to Chimera and wasn't really sure if there would be a reply (as has happened with some other MD products)
So thanks for your prompt response.
I had no problems with Build Structure after being pointed in the right direction. I will try text editor eventually just for the experience. I have successfully substituted S for O in a guanine complex, and will extend that to a Se substitution later.
The MD simulation runs much faster if I exclude the Periodic Boundary Conditions option. Is there a way to extend the run-time? Currently it covers 1000fs. Is getting to picosecond region a possibility?
Best wishes and congrats to whoever put Chimera MD together! Simon
On 28 December 2017 at 17:09, Elaine Meng <meng@cgl.ucsf.edu> wrote: Hi Simon, It would have been helpful if you said how you substituted S for O and what the error messages were. I have no idea what you did.
You can change atom type and element inside of Chimera or use a text editor (not in Chimera) to change the PDB file before opening it in Chimera.
The way to change the atom inside of Chimera is to select the atom and then use Build Structure (in menu under Tools… Structure Editing), the Modify Structure section. Use the option to change the name of the residue since if you keep the name the same, Chimera is expecting that residue, not something different with a sulphur in it. <http://www.rbvi.ucsf.edu/chimera/docs/ContributedSoftware/editing/editing.ht...>
If you try text-editing instead, remember spacing is important in PDB files, so don’t change the spacing. There's a summary of PDB format here: <http://www.rbvi.ucsf.edu/chimera/docs/UsersGuide/tutorials/framepdbintro.htm...>
In the text-editor of your choice, in the ATOM line for that atom, I would change the atom name, the element symbol (if present, would be near the end of the line), and in the ATOM lines for the whole residue, change the residue name.
Regardless of how you change the atom, however, another problem is that this nonstandard residue must be parametrized if you are going to run MD. Chimera will try to do this automatically using AMBER’s Antechamber module, but especially with highly charged residues such as nucleotides it may fail. In that case, I don’t really have any solution other than to try the simpler Gasteiger charges if it gives you a choice of Gasteiger or AM1-BCC. <http://www.rbvi.ucsf.edu/chimera/docs/ContributedSoftware/addcharge/addcharg...>
I hope this helps, Elaine ----- Elaine C. Meng, Ph.D. UCSF Chimera(X) team Department of Pharmaceutical Chemistry University of California, San Francisco
On Dec 27, 2017, at 1:49 PM, simon chapman <rowanlodge19@gmail.com> wrote:
Hello...for my Master's dissertation, I need to substitute a sulphur atom for an oxygen in a Molecular Dynamics simulation. The run works fine with my target molecule 1KF1.pdb uploaded into Chimera, but the substituted version sends quite a few error messages and won't run. It doesn't appear to recognise the novel S atom.
Any help would be much appreciated...I've been stuck on this for nearly a week!
Best wishes Simon
_______________________________________________ Chimera-users mailing list: Chimera-users@cgl.ucsf.edu Manage subscription: http://plato.cgl.ucsf.edu/mailman/listinfo/chimera-users
Very kind Elaine.Many thanks. I found a route to atomic substitution after a bit of tinkering. Tools>structure edit>build structure>modify structure. I'm getting some great data now! Very pleased. I will try your suggestions re extending the run-time. I don't really need anything more than a few ps at moment: my hypothesis is that a guanine quadruplex will be destabilised by incremental substitution at O6. So far the data are going my way! AMBER gave me real headaches...I have it set up in UBUNTU but on a Win10 machine, and the commands get a bit 'sticky'. It rather feels like the package has been poorly ported into UBUNTU in several stages over a long period by different designers. Also an over-detailed guide doesn't help. GROMACS is another LINUX-based system. NAMD works reasonably well, but Chimera tops the list so far. It will be a subsection of my dissertation. Best wishes Simon (Looks like you and I are the only folk working on New Year's Day!) On 1 January 2018 at 19:33, Elaine Meng <meng@cgl.ucsf.edu> wrote:
Hi Simon, The calculation runs on your own computer and as far as I know, there isn’t a limit on the run time. The “Run Parameters” section of the Molecular Dynamics SImulation dialog allows you to enter the numbers of steps for minimization, equilibration, and production. If you click on “minimization” you can see it is actually a little a menu that can switch to “equilibration” and “production” subsections.
<http://www.rbvi.ucsf.edu/chimera/docs/ContributedSoftware/md/md.html#run>
You can also specify a restart file for the production run, for subsequently doing another production run starting from the endpoint of the current one. As far as I know, there isn’t an upper limit to the number of steps per run, but it might be wiser to do multiple production runs anyway (so that if something happens, you don’t lose so much prior computation).
The tool was intended more to make MD simulations accessible to many people without too steep of a learning curve, rather than for very large-scale, long simulations which might be accomplished more efficiently with a dedicated MD package (AMBER, GROMACS, etc.), but you can certainly try.
I hope this helps, Elaine ----- Elaine C. Meng, Ph.D. UCSF Chimera(X) team Department of Pharmaceutical Chemistry University of California, San Francisco
On Dec 30, 2017, at 4:10 AM, simon chapman <rowanlodge19@gmail.com> wrote:
Hello Elaine. Yes,remiss of me to leave out so much info! I'm new-ish to Chimera and wasn't really sure if there would be a reply (as has happened with some other MD products)
So thanks for your prompt response.
I had no problems with Build Structure after being pointed in the right direction. I will try text editor eventually just for the experience. I have successfully substituted S for O in a guanine complex, and will extend that to a Se substitution later.
The MD simulation runs much faster if I exclude the Periodic Boundary Conditions option. Is there a way to extend the run-time? Currently it covers 1000fs. Is getting to picosecond region a possibility?
Best wishes and congrats to whoever put Chimera MD together! Simon
On 28 December 2017 at 17:09, Elaine Meng <meng@cgl.ucsf.edu> wrote: Hi Simon, It would have been helpful if you said how you substituted S for O and what the error messages were. I have no idea what you did.
You can change atom type and element inside of Chimera or use a text editor (not in Chimera) to change the PDB file before opening it in Chimera.
The way to change the atom inside of Chimera is to select the atom and then use Build Structure (in menu under Tools… Structure Editing), the Modify Structure section. Use the option to change the name of the residue since if you keep the name the same, Chimera is expecting that residue, not something different with a sulphur in it. <http://www.rbvi.ucsf.edu/chimera/docs/ContributedSoftware/editing/ editing.html#modify>
If you try text-editing instead, remember spacing is important in PDB files, so don’t change the spacing. There's a summary of PDB format here: <http://www.rbvi.ucsf.edu/chimera/docs/UsersGuide/ tutorials/framepdbintro.html>
In the text-editor of your choice, in the ATOM line for that atom, I would change the atom name, the element symbol (if present, would be near the end of the line), and in the ATOM lines for the whole residue, change the residue name.
Regardless of how you change the atom, however, another problem is that this nonstandard residue must be parametrized if you are going to run MD. Chimera will try to do this automatically using AMBER’s Antechamber module, but especially with highly charged residues such as nucleotides it may fail. In that case, I don’t really have any solution other than to try the simpler Gasteiger charges if it gives you a choice of Gasteiger or AM1-BCC. <http://www.rbvi.ucsf.edu/chimera/docs/ContributedSoftware/addcharge/ addcharge.html#antechamber>
I hope this helps, Elaine ----- Elaine C. Meng, Ph.D. UCSF Chimera(X) team Department of Pharmaceutical Chemistry University of California, San Francisco
On Dec 27, 2017, at 1:49 PM, simon chapman <rowanlodge19@gmail.com> wrote:
Hello...for my Master's dissertation, I need to substitute a sulphur atom for an oxygen in a Molecular Dynamics simulation. The run works fine with my target molecule 1KF1.pdb uploaded into Chimera, but the substituted version sends quite a few error messages and won't run. It doesn't appear to recognise the novel S atom.
Any help would be much appreciated...I've been stuck on this for nearly a week!
Best wishes Simon
_______________________________________________ Chimera-users mailing list: Chimera-users@cgl.ucsf.edu Manage subscription: http://plato.cgl.ucsf.edu/ mailman/listinfo/chimera-users
Hello Elaine, sorry to bother you again. Chimera has been a real treat so far, but I have hit a problem. I've struggled for a week now, and finally given up! I am incrementally substituting S for O on four guanines in a regular complex. 3 substitutions work fine in a simulation and producing very encouraging data (attachment 1) But 4 substitutions (attachment 2) triggers a load of error messages. The first is attachment 3. After solvation, attachment 4 appears and the solvation box is removed if neutralising ions are added. If ions not added, attachment 5 warning appears. Not sure if attachment 6 is relevant. Hope you can help/advise? Best wishes Simon On 1 January 2018 at 19:33, Elaine Meng <meng@cgl.ucsf.edu> wrote:
Hi Simon, The calculation runs on your own computer and as far as I know, there isn’t a limit on the run time. The “Run Parameters” section of the Molecular Dynamics SImulation dialog allows you to enter the numbers of steps for minimization, equilibration, and production. If you click on “minimization” you can see it is actually a little a menu that can switch to “equilibration” and “production” subsections.
<http://www.rbvi.ucsf.edu/chimera/docs/ContributedSoftware/md/md.html#run>
You can also specify a restart file for the production run, for subsequently doing another production run starting from the endpoint of the current one. As far as I know, there isn’t an upper limit to the number of steps per run, but it might be wiser to do multiple production runs anyway (so that if something happens, you don’t lose so much prior computation).
The tool was intended more to make MD simulations accessible to many people without too steep of a learning curve, rather than for very large-scale, long simulations which might be accomplished more efficiently with a dedicated MD package (AMBER, GROMACS, etc.), but you can certainly try.
I hope this helps, Elaine ----- Elaine C. Meng, Ph.D. UCSF Chimera(X) team Department of Pharmaceutical Chemistry University of California, San Francisco
On Dec 30, 2017, at 4:10 AM, simon chapman <rowanlodge19@gmail.com> wrote:
Hello Elaine. Yes,remiss of me to leave out so much info! I'm new-ish to Chimera and wasn't really sure if there would be a reply (as has happened with some other MD products)
So thanks for your prompt response.
I had no problems with Build Structure after being pointed in the right direction. I will try text editor eventually just for the experience. I have successfully substituted S for O in a guanine complex, and will extend that to a Se substitution later.
The MD simulation runs much faster if I exclude the Periodic Boundary Conditions option. Is there a way to extend the run-time? Currently it covers 1000fs. Is getting to picosecond region a possibility?
Best wishes and congrats to whoever put Chimera MD together! Simon
On 28 December 2017 at 17:09, Elaine Meng <meng@cgl.ucsf.edu> wrote: Hi Simon, It would have been helpful if you said how you substituted S for O and what the error messages were. I have no idea what you did.
You can change atom type and element inside of Chimera or use a text editor (not in Chimera) to change the PDB file before opening it in Chimera.
The way to change the atom inside of Chimera is to select the atom and then use Build Structure (in menu under Tools… Structure Editing), the Modify Structure section. Use the option to change the name of the residue since if you keep the name the same, Chimera is expecting that residue, not something different with a sulphur in it. <http://www.rbvi.ucsf.edu/chimera/docs/ContributedSoftware/editing/ editing.html#modify>
If you try text-editing instead, remember spacing is important in PDB files, so don’t change the spacing. There's a summary of PDB format here: <http://www.rbvi.ucsf.edu/chimera/docs/UsersGuide/ tutorials/framepdbintro.html>
In the text-editor of your choice, in the ATOM line for that atom, I would change the atom name, the element symbol (if present, would be near the end of the line), and in the ATOM lines for the whole residue, change the residue name.
Regardless of how you change the atom, however, another problem is that this nonstandard residue must be parametrized if you are going to run MD. Chimera will try to do this automatically using AMBER’s Antechamber module, but especially with highly charged residues such as nucleotides it may fail. In that case, I don’t really have any solution other than to try the simpler Gasteiger charges if it gives you a choice of Gasteiger or AM1-BCC. <http://www.rbvi.ucsf.edu/chimera/docs/ContributedSoftware/addcharge/ addcharge.html#antechamber>
I hope this helps, Elaine ----- Elaine C. Meng, Ph.D. UCSF Chimera(X) team Department of Pharmaceutical Chemistry University of California, San Francisco
On Dec 27, 2017, at 1:49 PM, simon chapman <rowanlodge19@gmail.com> wrote:
Hello...for my Master's dissertation, I need to substitute a sulphur atom for an oxygen in a Molecular Dynamics simulation. The run works fine with my target molecule 1KF1.pdb uploaded into Chimera, but the substituted version sends quite a few error messages and won't run. It doesn't appear to recognise the novel S atom.
Any help would be much appreciated...I've been stuck on this for nearly a week!
Best wishes Simon
_______________________________________________ Chimera-users mailing list: Chimera-users@cgl.ucsf.edu Manage subscription: http://plato.cgl.ucsf.edu/ mailman/listinfo/chimera-users
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
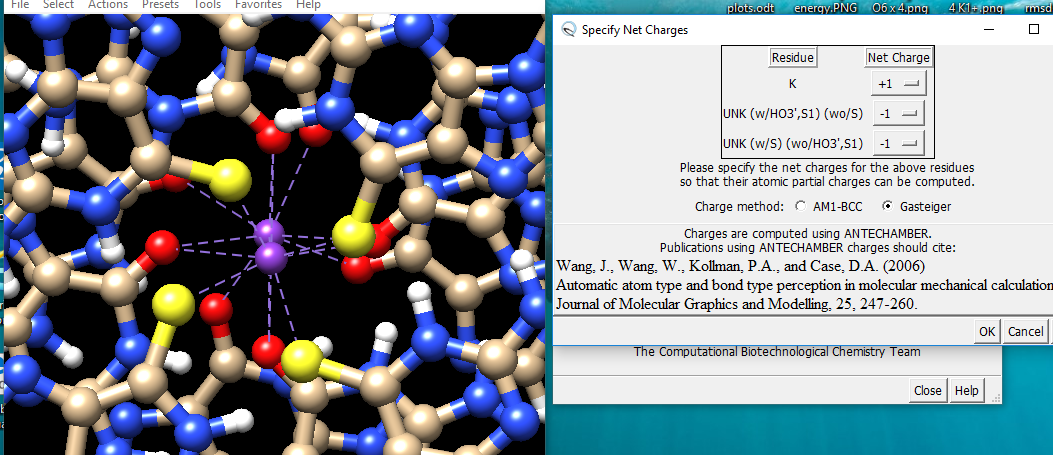
Hi Simon, I think you may be stretching the tools beyond their intended purposes: Chimera’s minimization, molecular dynamics interface, and automatic parametrization of nonstandard residues with Antechamber are meant to provide relatively simple access to such calculations by noncomputational scientists, given the large learning curves of dedicated simulation packages like GROMACS, AMBER, etc. These tools can be highly useful to clean up structures or suggest additional reasonable conformations. However, they are not meant for precise quantification of energies or for very long simulations, especially not in conjunction with nonstandard residues with parameters estimated by Antechamber. The Gasteiger charges are very quick-and-dirty, so I would caution against overinterpreting simulations that employ them. Further, Antechember is meant to cover most small organic molecules but not every possible molecule. It seems that the modified UNK residues fall into two structural types (as per the “specify net charges” dialog), and that one (maybe just the 4th?) fails in parametrization by Antechamber, and then the Solvate or Add Ions tool cannot find some parameter needed for its calculation. These tools come from the Ambertools package even though they are included with Chimera and Chimera has interfaces to them. I don’t know exactly what the problem is, nor do I have a solution, sorry. Elaine ----- Elaine C. Meng, Ph.D. UCSF Chimera(X) team Department of Pharmaceutical Chemistry University of California, San Francisco
On Jan 9, 2018, at 1:30 AM, simon chapman <rowanlodge19@gmail.com> wrote:
Hello Elaine, sorry to bother you again.
Chimera has been a real treat so far, but I have hit a problem. I've struggled for a week now, and finally given up!
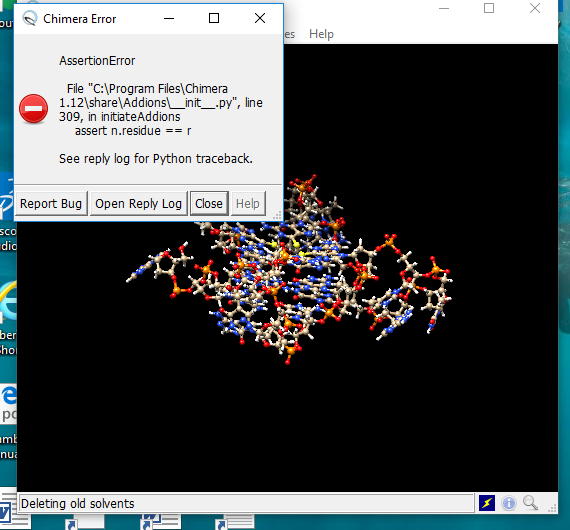
I am incrementally substituting S for O on four guanines in a regular complex. 3 substitutions work fine in a simulation and producing very encouraging data (attachment 1) But 4 substitutions (attachment 2) triggers a load of error messages. The first is attachment 3. After solvation, attachment 4 appears and the solvation box is removed if neutralising ions are added. If ions not added, attachment 5 warning appears. Not sure if attachment 6 is relevant.
Hope you can help/advise?
Best wishes Simon

Hi Simon, Everything Elaine says is true. I do have some last ditch suggestions for you though. One thing is that the fourth guanine differs in protonation state from the others in that the fourth one either has or lacks an HO3’ whereas the others don’t. You should investigate why this is the case. The two possibilities are then: 1) The protonation difference is an error. You would need to correct the structure so that all four guanines start with the same protonation. 2) For some reason the protonation difference is okay. You then need to give the modified version of the fourth guanine a different name from the others, e.g. “UK2” instead of “UNK”. Not guaranteeing this will work, but it might. —Eric Eric Pettersen UCSF Computer Graphics Lab On Jan 9, 2018, at 10:24 AM, Elaine Meng <meng@cgl.ucsf.edu> wrote:
Hi Simon, I think you may be stretching the tools beyond their intended purposes: Chimera’s minimization, molecular dynamics interface, and automatic parametrization of nonstandard residues with Antechamber are meant to provide relatively simple access to such calculations by noncomputational scientists, given the large learning curves of dedicated simulation packages like GROMACS, AMBER, etc. These tools can be highly useful to clean up structures or suggest additional reasonable conformations. However, they are not meant for precise quantification of energies or for very long simulations, especially not in conjunction with nonstandard residues with parameters estimated by Antechamber. The Gasteiger charges are very quick-and-dirty, so I would caution against overinterpreting simulations that employ them. Further, Antechember is meant to cover most small organic molecules but not every possible molecule.
It seems that the modified UNK residues fall into two structural types (as per the “specify net charges” dialog), and that one (maybe just the 4th?) fails in parametrization by Antechamber, and then the Solvate or Add Ions tool cannot find some parameter needed for its calculation. These tools come from the Ambertools package even though they are included with Chimera and Chimera has interfaces to them. I don’t know exactly what the problem is, nor do I have a solution, sorry. Elaine ----- Elaine C. Meng, Ph.D. UCSF Chimera(X) team Department of Pharmaceutical Chemistry University of California, San Francisco
On Jan 9, 2018, at 1:30 AM, simon chapman <rowanlodge19@gmail.com> wrote:
Hello Elaine, sorry to bother you again.
Chimera has been a real treat so far, but I have hit a problem. I've struggled for a week now, and finally given up!
I am incrementally substituting S for O on four guanines in a regular complex. 3 substitutions work fine in a simulation and producing very encouraging data (attachment 1) But 4 substitutions (attachment 2) triggers a load of error messages. The first is attachment 3. After solvation, attachment 4 appears and the solvation box is removed if neutralising ions are added. If ions not added, attachment 5 warning appears. Not sure if attachment 6 is relevant.
Hope you can help/advise?
Best wishes Simon
_______________________________________________ Chimera-users mailing list: Chimera-users@cgl.ucsf.edu Manage subscription: http://plato.cgl.ucsf.edu/mailman/listinfo/chimera-users
Hello Elaine and Eric. Thankyou for the prompt reply....and I'm very impressed! Eric's suggestion #2 worked! I assumed the protonation was OK as I ran the PDB downloaded structure through a protonation webserver (H++). So I just changed the name of the novel substitution. It looks like additional alterations will also need renaming: I ran the next substitution in the incremental progression( #5) as "UNK3",and that is just about to complete. I am especially pleased as the data are definitely tending to support my hypothesis. You will acknowledged in the Methodology section of my dissertation. So, thanks again. There's a charming 14th century pub just down the lane from me...buy you both a pint next time you're passing through Best wishes Simon On 9 January 2018 at 19:31, Eric Pettersen <pett@cgl.ucsf.edu> wrote:
Hi Simon, Everything Elaine says is true. I do have some last ditch suggestions for you though. One thing is that the fourth guanine differs in protonation state from the others in that the fourth one either has or lacks an HO3’ whereas the others don’t. You should investigate why this is the case. The two possibilities are then:
1) The protonation difference is an error. You would need to correct the structure so that all four guanines start with the same protonation. 2) For some reason the protonation difference is okay. You then need to give the modified version of the fourth guanine a different name from the others, e.g. “UK2” instead of “UNK”. Not guaranteeing this will work, but it might.
—Eric
Eric Pettersen UCSF Computer Graphics Lab
On Jan 9, 2018, at 10:24 AM, Elaine Meng <meng@cgl.ucsf.edu> wrote:
Hi Simon, I think you may be stretching the tools beyond their intended purposes: Chimera’s minimization, molecular dynamics interface, and automatic parametrization of nonstandard residues with Antechamber are meant to provide relatively simple access to such calculations by noncomputational scientists, given the large learning curves of dedicated simulation packages like GROMACS, AMBER, etc. These tools can be highly useful to clean up structures or suggest additional reasonable conformations. However, they are not meant for precise quantification of energies or for very long simulations, especially not in conjunction with nonstandard residues with parameters estimated by Antechamber. The Gasteiger charges are very quick-and-dirty, so I would caution against overinterpreting simulations that employ them. Further, Antechember is meant to cover most small organic molecules but not every possible molecule.
It seems that the modified UNK residues fall into two structural types (as per the “specify net charges” dialog), and that one (maybe just the 4th?) fails in parametrization by Antechamber, and then the Solvate or Add Ions tool cannot find some parameter needed for its calculation. These tools come from the Ambertools package even though they are included with Chimera and Chimera has interfaces to them. I don’t know exactly what the problem is, nor do I have a solution, sorry. Elaine ----- Elaine C. Meng, Ph.D. UCSF Chimera(X) team Department of Pharmaceutical Chemistry University of California, San Francisco
On Jan 9, 2018, at 1:30 AM, simon chapman <rowanlodge19@gmail.com> wrote:
Hello Elaine, sorry to bother you again.
Chimera has been a real treat so far, but I have hit a problem. I've struggled for a week now, and finally given up!
I am incrementally substituting S for O on four guanines in a regular complex. 3 substitutions work fine in a simulation and producing very encouraging data (attachment 1) But 4 substitutions (attachment 2) triggers a load of error messages. The first is attachment 3. After solvation, attachment 4 appears and the solvation box is removed if neutralising ions are added. If ions not added, attachment 5 warning appears. Not sure if attachment 6 is relevant.
Hope you can help/advise?
Best wishes Simon
_______________________________________________ Chimera-users mailing list: Chimera-users@cgl.ucsf.edu Manage subscription: http://plato.cgl.ucsf.edu/ mailman/listinfo/chimera-users
Great! If we’re in the neighborhood I’ll take a pint of ginger ale. :-) —Eric
On Jan 10, 2018, at 4:09 AM, simon chapman <rowanlodge19@gmail.com> wrote:
Hello Elaine and Eric. Thankyou for the prompt reply....and I'm very impressed! Eric's suggestion #2 worked!
I assumed the protonation was OK as I ran the PDB downloaded structure through a protonation webserver (H++). So I just changed the name of the novel substitution. It looks like additional alterations will also need renaming: I ran the next substitution in the incremental progression( #5) as "UNK3",and that is just about to complete.
I am especially pleased as the data are definitely tending to support my hypothesis. You will acknowledged in the Methodology section of my dissertation.
So, thanks again. There's a charming 14th century pub just down the lane from me...buy you both a pint next time you're passing through
Best wishes Simon
On 9 January 2018 at 19:31, Eric Pettersen <pett@cgl.ucsf.edu <mailto:pett@cgl.ucsf.edu>> wrote: Hi Simon, Everything Elaine says is true. I do have some last ditch suggestions for you though. One thing is that the fourth guanine differs in protonation state from the others in that the fourth one either has or lacks an HO3’ whereas the others don’t. You should investigate why this is the case. The two possibilities are then:
1) The protonation difference is an error. You would need to correct the structure so that all four guanines start with the same protonation. 2) For some reason the protonation difference is okay. You then need to give the modified version of the fourth guanine a different name from the others, e.g. “UK2” instead of “UNK”. Not guaranteeing this will work, but it might.
—Eric
Eric Pettersen UCSF Computer Graphics Lab
On Jan 9, 2018, at 10:24 AM, Elaine Meng <meng@cgl.ucsf.edu <mailto:meng@cgl.ucsf.edu>> wrote:
Hi Simon, I think you may be stretching the tools beyond their intended purposes: Chimera’s minimization, molecular dynamics interface, and automatic parametrization of nonstandard residues with Antechamber are meant to provide relatively simple access to such calculations by noncomputational scientists, given the large learning curves of dedicated simulation packages like GROMACS, AMBER, etc. These tools can be highly useful to clean up structures or suggest additional reasonable conformations. However, they are not meant for precise quantification of energies or for very long simulations, especially not in conjunction with nonstandard residues with parameters estimated by Antechamber. The Gasteiger charges are very quick-and-dirty, so I would caution against overinterpreting simulations that employ them. Further, Antechember is meant to cover most small organic molecules but not every possible molecule.
It seems that the modified UNK residues fall into two structural types (as per the “specify net charges” dialog), and that one (maybe just the 4th?) fails in parametrization by Antechamber, and then the Solvate or Add Ions tool cannot find some parameter needed for its calculation. These tools come from the Ambertools package even though they are included with Chimera and Chimera has interfaces to them. I don’t know exactly what the problem is, nor do I have a solution, sorry. Elaine ----- Elaine C. Meng, Ph.D. UCSF Chimera(X) team Department of Pharmaceutical Chemistry University of California, San Francisco
On Jan 9, 2018, at 1:30 AM, simon chapman <rowanlodge19@gmail.com <mailto:rowanlodge19@gmail.com>> wrote:
Hello Elaine, sorry to bother you again.
Chimera has been a real treat so far, but I have hit a problem. I've struggled for a week now, and finally given up!
I am incrementally substituting S for O on four guanines in a regular complex. 3 substitutions work fine in a simulation and producing very encouraging data (attachment 1) But 4 substitutions (attachment 2) triggers a load of error messages. The first is attachment 3. After solvation, attachment 4 appears and the solvation box is removed if neutralising ions are added. If ions not added, attachment 5 warning appears. Not sure if attachment 6 is relevant.
Hope you can help/advise?
Best wishes Simon
_______________________________________________ Chimera-users mailing list: Chimera-users@cgl.ucsf.edu <mailto:Chimera-users@cgl.ucsf.edu> Manage subscription: http://plato.cgl.ucsf.edu/mailman/listinfo/chimera-users <http://plato.cgl.ucsf.edu/mailman/listinfo/chimera-users>
Hello Eric and Elaine. Chance of earning a second pint of ginger beer if you wish... Simulations and analysis going very well, I have substituted all 12 oxygens for sulphur. Each requires a new name: UNK I, UNK2 etc. Advice for others...naming will only alow digits 1 to 9. so for 12 iterations, I used zero and the original unappended "UNK". Which makes 12. I have also successfully set production for 50000steps, equivalent to 8ps. However, there appears to be an option for continuation from one run to a subsequent one. I've tried for a few days now, but get the same error message every time ('continuation2 attachment'). Changing file names as described in the message doesn't work. It's probably something simple (probably me in fact), but would really appreciate your comments. Best wishes Simon On 10 January 2018 at 19:01, Eric Pettersen <pett@cgl.ucsf.edu> wrote:
Great! If we’re in the neighborhood I’ll take a pint of ginger ale. :-)
—Eric
On Jan 10, 2018, at 4:09 AM, simon chapman <rowanlodge19@gmail.com> wrote:
Hello Elaine and Eric. Thankyou for the prompt reply....and I'm very impressed! Eric's suggestion #2 worked!
I assumed the protonation was OK as I ran the PDB downloaded structure through a protonation webserver (H++). So I just changed the name of the novel substitution. It looks like additional alterations will also need renaming: I ran the next substitution in the incremental progression( #5) as "UNK3",and that is just about to complete.
I am especially pleased as the data are definitely tending to support my hypothesis. You will acknowledged in the Methodology section of my dissertation.
So, thanks again. There's a charming 14th century pub just down the lane from me...buy you both a pint next time you're passing through
Best wishes Simon
On 9 January 2018 at 19:31, Eric Pettersen <pett@cgl.ucsf.edu> wrote:
Hi Simon, Everything Elaine says is true. I do have some last ditch suggestions for you though. One thing is that the fourth guanine differs in protonation state from the others in that the fourth one either has or lacks an HO3’ whereas the others don’t. You should investigate why this is the case. The two possibilities are then:
1) The protonation difference is an error. You would need to correct the structure so that all four guanines start with the same protonation. 2) For some reason the protonation difference is okay. You then need to give the modified version of the fourth guanine a different name from the others, e.g. “UK2” instead of “UNK”. Not guaranteeing this will work, but it might.
—Eric
Eric Pettersen UCSF Computer Graphics Lab
On Jan 9, 2018, at 10:24 AM, Elaine Meng <meng@cgl.ucsf.edu> wrote:
Hi Simon, I think you may be stretching the tools beyond their intended purposes: Chimera’s minimization, molecular dynamics interface, and automatic parametrization of nonstandard residues with Antechamber are meant to provide relatively simple access to such calculations by noncomputational scientists, given the large learning curves of dedicated simulation packages like GROMACS, AMBER, etc. These tools can be highly useful to clean up structures or suggest additional reasonable conformations. However, they are not meant for precise quantification of energies or for very long simulations, especially not in conjunction with nonstandard residues with parameters estimated by Antechamber. The Gasteiger charges are very quick-and-dirty, so I would caution against overinterpreting simulations that employ them. Further, Antechember is meant to cover most small organic molecules but not every possible molecule.
It seems that the modified UNK residues fall into two structural types (as per the “specify net charges” dialog), and that one (maybe just the 4th?) fails in parametrization by Antechamber, and then the Solvate or Add Ions tool cannot find some parameter needed for its calculation. These tools come from the Ambertools package even though they are included with Chimera and Chimera has interfaces to them. I don’t know exactly what the problem is, nor do I have a solution, sorry. Elaine ----- Elaine C. Meng, Ph.D. UCSF Chimera(X) team Department of Pharmaceutical Chemistry University of California, San Francisco
On Jan 9, 2018, at 1:30 AM, simon chapman <rowanlodge19@gmail.com> wrote:
Hello Elaine, sorry to bother you again.
Chimera has been a real treat so far, but I have hit a problem. I've struggled for a week now, and finally given up!
I am incrementally substituting S for O on four guanines in a regular complex. 3 substitutions work fine in a simulation and producing very encouraging data (attachment 1) But 4 substitutions (attachment 2) triggers a load of error messages. The first is attachment 3. After solvation, attachment 4 appears and the solvation box is removed if neutralising ions are added. If ions not added, attachment 5 warning appears. Not sure if attachment 6 is relevant.
Hope you can help/advise?
Best wishes Simon
_______________________________________________ Chimera-users mailing list: Chimera-users@cgl.ucsf.edu Manage subscription: http://plato.cgl.ucsf.edu/mail man/listinfo/chimera-users
{kind=link}
{kind=link}
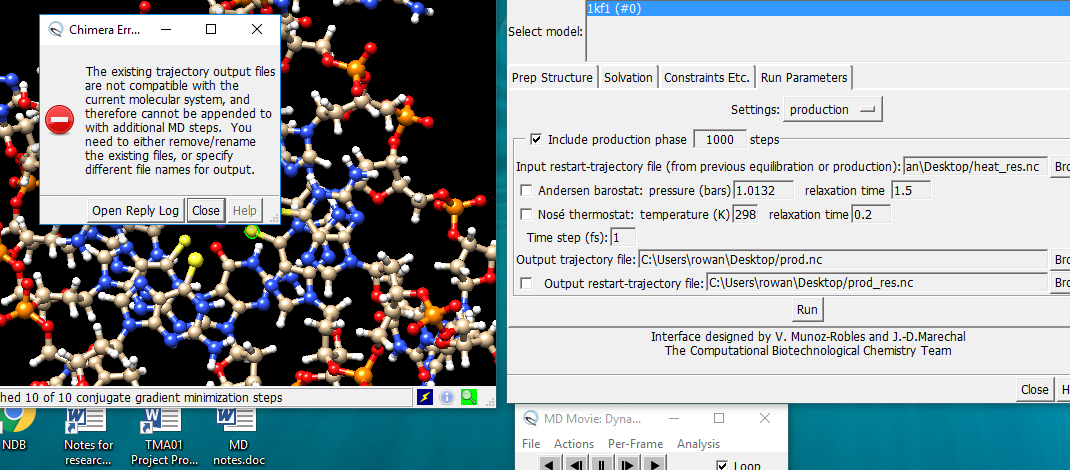
As I understand it, the message is that the molecules in the input restart file are not the same as the current molecules (not exactly the same set of residues, atoms, bonds). Changing the filenames would allow for a completely separate run, but it would not solve the problem of continuing the same simulation if the chemical system is different. How it might be different, I don’t know. All I can say is if you changed something between the runs, don’t do that. Elaine
On Jan 17, 2018, at 4:33 AM, simon chapman <rowanlodge19@gmail.com> wrote:
Hello Eric and Elaine. Chance of earning a second pint of ginger beer if you wish...
Simulations and analysis going very well, I have substituted all 12 oxygens for sulphur. Each requires a new name: UNK I, UNK2 etc. Advice for others...naming will only alow digits 1 to 9. so for 12 iterations, I used zero and the original unappended "UNK". Which makes 12. I have also successfully set production for 50000steps, equivalent to 8ps.
However, there appears to be an option for continuation from one run to a subsequent one. I've tried for a few days now, but get the same error message every time ('continuation2 attachment'). Changing file names as described in the message doesn't work. It's probably something simple (probably me in fact), but would really appreciate your comments.
Best wishes Simon
On 10 January 2018 at 19:01, Eric Pettersen <pett@cgl.ucsf.edu> wrote: Great! If we’re in the neighborhood I’ll take a pint of ginger ale. :-)
—Eric
On Jan 10, 2018, at 4:09 AM, simon chapman <rowanlodge19@gmail.com> wrote:
Hello Elaine and Eric. Thankyou for the prompt reply....and I'm very impressed! Eric's suggestion #2 worked!
I assumed the protonation was OK as I ran the PDB downloaded structure through a protonation webserver (H++). So I just changed the name of the novel substitution. It looks like additional alterations will also need renaming: I ran the next substitution in the incremental progression( #5) as "UNK3",and that is just about to complete.
I am especially pleased as the data are definitely tending to support my hypothesis. You will acknowledged in the Methodology section of my dissertation.
So, thanks again. There's a charming 14th century pub just down the lane from me...buy you both a pint next time you're passing through
Best wishes Simon
On 9 January 2018 at 19:31, Eric Pettersen <pett@cgl.ucsf.edu> wrote: Hi Simon, Everything Elaine says is true. I do have some last ditch suggestions for you though. One thing is that the fourth guanine differs in protonation state from the others in that the fourth one either has or lacks an HO3’ whereas the others don’t. You should investigate why this is the case. The two possibilities are then:
1) The protonation difference is an error. You would need to correct the structure so that all four guanines start with the same protonation. 2) For some reason the protonation difference is okay. You then need to give the modified version of the fourth guanine a different name from the others, e.g. “UK2” instead of “UNK”. Not guaranteeing this will work, but it might.
—Eric
Eric Pettersen UCSF Computer Graphics Lab
On Jan 9, 2018, at 10:24 AM, Elaine Meng <meng@cgl.ucsf.edu> wrote:
Hi Simon, I think you may be stretching the tools beyond their intended purposes: Chimera’s minimization, molecular dynamics interface, and automatic parametrization of nonstandard residues with Antechamber are meant to provide relatively simple access to such calculations by noncomputational scientists, given the large learning curves of dedicated simulation packages like GROMACS, AMBER, etc. These tools can be highly useful to clean up structures or suggest additional reasonable conformations. However, they are not meant for precise quantification of energies or for very long simulations, especially not in conjunction with nonstandard residues with parameters estimated by Antechamber. The Gasteiger charges are very quick-and-dirty, so I would caution against overinterpreting simulations that employ them. Further, Antechember is meant to cover most small organic molecules but not every possible molecule.
It seems that the modified UNK residues fall into two structural types (as per the “specify net charges” dialog), and that one (maybe just the 4th?) fails in parametrization by Antechamber, and then the Solvate or Add Ions tool cannot find some parameter needed for its calculation. These tools come from the Ambertools package even though they are included with Chimera and Chimera has interfaces to them. I don’t know exactly what the problem is, nor do I have a solution, sorry. Elaine ----- Elaine C. Meng, Ph.D. UCSF Chimera(X) team Department of Pharmaceutical Chemistry University of California, San Francisco
On Jan 9, 2018, at 1:30 AM, simon chapman <rowanlodge19@gmail.com> wrote:
Hello Elaine, sorry to bother you again.
Chimera has been a real treat so far, but I have hit a problem. I've struggled for a week now, and finally given up!
I am incrementally substituting S for O on four guanines in a regular complex. 3 substitutions work fine in a simulation and producing very encouraging data (attachment 1) But 4 substitutions (attachment 2) triggers a load of error messages. The first is attachment 3. After solvation, attachment 4 appears and the solvation box is removed if neutralising ions are added. If ions not added, attachment 5 warning appears. Not sure if attachment 6 is relevant.
Hope you can help/advise?
Best wishes Simon
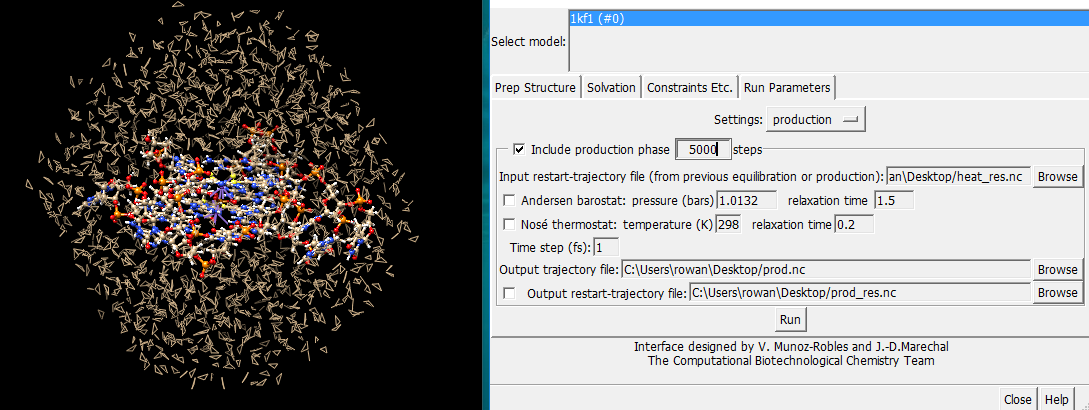
Hello Elaine and Eric...nothing to worry about, just updating you with some useful info. The difficulties I had with continuation runs in Chimera took several weeks to resolve, but now I can get to ~200ps as opposed to the 5ps value of a standard run. The problem lies in the Prep Structure interface (attached). The instruction "Input restart-trajectory file from previous equilibration/production" would reasonably be interpreted as inputting the restart-trajectory file from an earlier run? This file is generated after each run and is posted on the desktop. However, it's never accepted, and the error message I mentioned in an earlier email pops up every time. It actually needs to have a suffix <01. Which is very odd,as such a file doesn't actually exist until the run is over! Adding a suffixes of unit value higher to the output files is however logical. It might be just ambiguous interface design, something that slipped thru the net, but the problem is very simply resolved by this method. If only it hadn't taken me weeks to stumble on it! Still very happy with Chimera, getting excellent data for my dissertation. Best wishes Simon On 17 January 2018 at 18:38, Elaine Meng <meng@cgl.ucsf.edu> wrote:
As I understand it, the message is that the molecules in the input restart file are not the same as the current molecules (not exactly the same set of residues, atoms, bonds). Changing the filenames would allow for a completely separate run, but it would not solve the problem of continuing the same simulation if the chemical system is different. How it might be different, I don’t know. All I can say is if you changed something between the runs, don’t do that. Elaine
On Jan 17, 2018, at 4:33 AM, simon chapman <rowanlodge19@gmail.com> wrote:
Hello Eric and Elaine. Chance of earning a second pint of ginger beer if you wish...
Simulations and analysis going very well, I have substituted all 12 oxygens for sulphur. Each requires a new name: UNK I, UNK2 etc. Advice for others...naming will only alow digits 1 to 9. so for 12 iterations, I used zero and the original unappended "UNK". Which makes 12. I have also successfully set production for 50000steps, equivalent to 8ps.
However, there appears to be an option for continuation from one run to a subsequent one. I've tried for a few days now, but get the same error message every time ('continuation2 attachment'). Changing file names as described in the message doesn't work. It's probably something simple (probably me in fact), but would really appreciate your comments.
Best wishes Simon
On 10 January 2018 at 19:01, Eric Pettersen <pett@cgl.ucsf.edu> wrote: Great! If we’re in the neighborhood I’ll take a pint of ginger ale. :-)
—Eric
On Jan 10, 2018, at 4:09 AM, simon chapman <rowanlodge19@gmail.com> wrote:
Hello Elaine and Eric. Thankyou for the prompt reply....and I'm very impressed! Eric's suggestion #2 worked!
I assumed the protonation was OK as I ran the PDB downloaded structure through a protonation webserver (H++). So I just changed the name of the novel substitution. It looks like additional alterations will also need renaming: I ran the next substitution in the incremental progression( #5) as "UNK3",and that is just about to complete.
I am especially pleased as the data are definitely tending to support my hypothesis. You will acknowledged in the Methodology section of my dissertation.
So, thanks again. There's a charming 14th century pub just down the lane from me...buy you both a pint next time you're passing through
Best wishes Simon
On 9 January 2018 at 19:31, Eric Pettersen <pett@cgl.ucsf.edu> wrote: Hi Simon, Everything Elaine says is true. I do have some last ditch suggestions for you though. One thing is that the fourth guanine differs in protonation state from the others in that the fourth one either has or lacks an HO3’ whereas the others don’t. You should investigate why this is the case. The two possibilities are then:
1) The protonation difference is an error. You would need to correct the structure so that all four guanines start with the same protonation. 2) For some reason the protonation difference is okay. You then need to give the modified version of the fourth guanine a different name from the others, e.g. “UK2” instead of “UNK”. Not guaranteeing this will work, but it might.
—Eric
Eric Pettersen UCSF Computer Graphics Lab
On Jan 9, 2018, at 10:24 AM, Elaine Meng <meng@cgl.ucsf.edu> wrote:
Hi Simon, I think you may be stretching the tools beyond their intended
purposes: Chimera’s minimization, molecular dynamics interface, and automatic parametrization of nonstandard residues with Antechamber are meant to provide relatively simple access to such calculations by noncomputational scientists, given the large learning curves of dedicated simulation packages like GROMACS, AMBER, etc. These tools can be highly useful to clean up structures or suggest additional reasonable conformations. However, they are not meant for precise quantification of energies or for very long simulations, especially not in conjunction with nonstandard residues with parameters estimated by Antechamber. The Gasteiger charges are very quick-and-dirty, so I would caution against overinterpreting simulations that employ them. Further, Antechember is meant to cover most small organic molecules but not every possible molecule.
It seems that the modified UNK residues fall into two structural types
(as per the “specify net charges” dialog), and that one (maybe just the 4th?) fails in parametrization by Antechamber, and then the Solvate or Add Ions tool cannot find some parameter needed for its calculation. These tools come from the Ambertools package even though they are included with Chimera and Chimera has interfaces to them. I don’t know exactly what the problem is, nor do I have a solution, sorry.
Elaine ----- Elaine C. Meng, Ph.D. UCSF Chimera(X) team Department of Pharmaceutical Chemistry University of California, San Francisco
On Jan 9, 2018, at 1:30 AM, simon chapman <rowanlodge19@gmail.com> wrote:
Hello Elaine, sorry to bother you again.
Chimera has been a real treat so far, but I have hit a problem. I've struggled for a week now, and finally given up!
I am incrementally substituting S for O on four guanines in a regular complex. 3 substitutions work fine in a simulation and producing very encouraging data (attachment 1) But 4 substitutions (attachment 2) triggers a load of error messages. The first is attachment 3. After solvation, attachment 4 appears and the solvation box is removed if neutralising ions are added. If ions not added, attachment 5 warning appears. Not sure if attachment 6 is relevant.
Hope you can help/advise?
Best wishes Simon
{kind=link}
Thanks Simon, that’s valuable information! We’ll try to document how that works. —Eric
On Mar 5, 2018, at 6:45 AM, simon chapman <rowanlodge19@gmail.com> wrote:
Hello Elaine and Eric...nothing to worry about, just updating you with some useful info.
The difficulties I had with continuation runs in Chimera took several weeks to resolve, but now I can get to ~200ps as opposed to the 5ps value of a standard run.
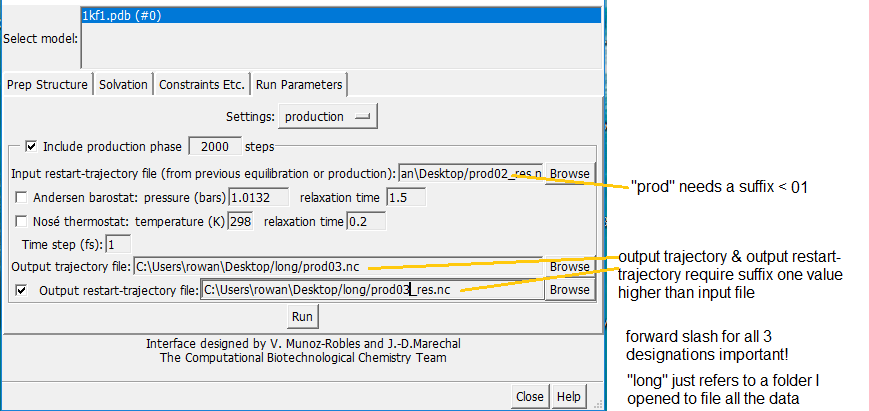
The problem lies in the Prep Structure interface (attached). The instruction "Input restart-trajectory file from previous equilibration/production" would reasonably be interpreted as inputting the restart-trajectory file from an earlier run? This file is generated after each run and is posted on the desktop. However, it's never accepted, and the error message I mentioned in an earlier email pops up every time.
It actually needs to have a suffix <01. Which is very odd,as such a file doesn't actually exist until the run is over! Adding a suffixes of unit value higher to the output files is however logical.
It might be just ambiguous interface design, something that slipped thru the net, but the problem is very simply resolved by this method. If only it hadn't taken me weeks to stumble on it!
Still very happy with Chimera, getting excellent data for my dissertation. Best wishes Simon
On 17 January 2018 at 18:38, Elaine Meng <meng@cgl.ucsf.edu <mailto:meng@cgl.ucsf.edu>> wrote: As I understand it, the message is that the molecules in the input restart file are not the same as the current molecules (not exactly the same set of residues, atoms, bonds). Changing the filenames would allow for a completely separate run, but it would not solve the problem of continuing the same simulation if the chemical system is different. How it might be different, I don’t know. All I can say is if you changed something between the runs, don’t do that. Elaine
On Jan 17, 2018, at 4:33 AM, simon chapman <rowanlodge19@gmail.com <mailto:rowanlodge19@gmail.com>> wrote:
Hello Eric and Elaine. Chance of earning a second pint of ginger beer if you wish...
Simulations and analysis going very well, I have substituted all 12 oxygens for sulphur. Each requires a new name: UNK I, UNK2 etc. Advice for others...naming will only alow digits 1 to 9. so for 12 iterations, I used zero and the original unappended "UNK". Which makes 12. I have also successfully set production for 50000steps, equivalent to 8ps.
However, there appears to be an option for continuation from one run to a subsequent one. I've tried for a few days now, but get the same error message every time ('continuation2 attachment'). Changing file names as described in the message doesn't work. It's probably something simple (probably me in fact), but would really appreciate your comments.
Best wishes Simon
On 10 January 2018 at 19:01, Eric Pettersen <pett@cgl.ucsf.edu <mailto:pett@cgl.ucsf.edu>> wrote: Great! If we’re in the neighborhood I’ll take a pint of ginger ale. :-)
—Eric
On Jan 10, 2018, at 4:09 AM, simon chapman <rowanlodge19@gmail.com <mailto:rowanlodge19@gmail.com>> wrote:
Hello Elaine and Eric. Thankyou for the prompt reply....and I'm very impressed! Eric's suggestion #2 worked!
I assumed the protonation was OK as I ran the PDB downloaded structure through a protonation webserver (H++). So I just changed the name of the novel substitution. It looks like additional alterations will also need renaming: I ran the next substitution in the incremental progression( #5) as "UNK3",and that is just about to complete.
I am especially pleased as the data are definitely tending to support my hypothesis. You will acknowledged in the Methodology section of my dissertation.
So, thanks again. There's a charming 14th century pub just down the lane from me...buy you both a pint next time you're passing through
Best wishes Simon
On 9 January 2018 at 19:31, Eric Pettersen <pett@cgl.ucsf.edu <mailto:pett@cgl.ucsf.edu>> wrote: Hi Simon, Everything Elaine says is true. I do have some last ditch suggestions for you though. One thing is that the fourth guanine differs in protonation state from the others in that the fourth one either has or lacks an HO3’ whereas the others don’t. You should investigate why this is the case. The two possibilities are then:
1) The protonation difference is an error. You would need to correct the structure so that all four guanines start with the same protonation. 2) For some reason the protonation difference is okay. You then need to give the modified version of the fourth guanine a different name from the others, e.g. “UK2” instead of “UNK”. Not guaranteeing this will work, but it might.
—Eric
Eric Pettersen UCSF Computer Graphics Lab
On Jan 9, 2018, at 10:24 AM, Elaine Meng <meng@cgl.ucsf.edu <mailto:meng@cgl.ucsf.edu>> wrote:
Hi Simon, I think you may be stretching the tools beyond their intended purposes: Chimera’s minimization, molecular dynamics interface, and automatic parametrization of nonstandard residues with Antechamber are meant to provide relatively simple access to such calculations by noncomputational scientists, given the large learning curves of dedicated simulation packages like GROMACS, AMBER, etc. These tools can be highly useful to clean up structures or suggest additional reasonable conformations. However, they are not meant for precise quantification of energies or for very long simulations, especially not in conjunction with nonstandard residues with parameters estimated by Antechamber. The Gasteiger charges are very quick-and-dirty, so I would caution against overinterpreting simulations that employ them. Further, Antechember is meant to cover most small organic molecules but not every possible molecule.
It seems that the modified UNK residues fall into two structural types (as per the “specify net charges” dialog), and that one (maybe just the 4th?) fails in parametrization by Antechamber, and then the Solvate or Add Ions tool cannot find some parameter needed for its calculation. These tools come from the Ambertools package even though they are included with Chimera and Chimera has interfaces to them. I don’t know exactly what the problem is, nor do I have a solution, sorry. Elaine ----- Elaine C. Meng, Ph.D. UCSF Chimera(X) team Department of Pharmaceutical Chemistry University of California, San Francisco
On Jan 9, 2018, at 1:30 AM, simon chapman <rowanlodge19@gmail.com <mailto:rowanlodge19@gmail.com>> wrote:
Hello Elaine, sorry to bother you again.
Chimera has been a real treat so far, but I have hit a problem. I've struggled for a week now, and finally given up!
I am incrementally substituting S for O on four guanines in a regular complex. 3 substitutions work fine in a simulation and producing very encouraging data (attachment 1) But 4 substitutions (attachment 2) triggers a load of error messages. The first is attachment 3. After solvation, attachment 4 appears and the solvation box is removed if neutralising ions are added. If ions not added, attachment 5 warning appears. Not sure if attachment 6 is relevant.
Hope you can help/advise?
Best wishes Simon
<prod phase for chimera team.png>_______________________________________________ Chimera-users mailing list: Chimera-users@cgl.ucsf.edu <mailto:Chimera-users@cgl.ucsf.edu> Manage subscription: http://plato.cgl.ucsf.edu/mailman/listinfo/chimera-users <http://plato.cgl.ucsf.edu/mailman/listinfo/chimera-users>
Hi Simon, Upon further review, I think you were just getting lucky here. There was a bug where systems with polymers in them might or might not match depending on basically random factors (the ordering of polymeric residues as given to MMTK — which were in no particular order). I have fixed the problem and think things will work now. The fix will be in tomorrow’s daily build (any daily build dated April 3 or later will have the fix). —Eric Eric Pettersen UCSF Computer Graphics Lab
On Mar 5, 2018, at 6:45 AM, simon chapman <rowanlodge19@gmail.com> wrote:
Hello Elaine and Eric...nothing to worry about, just updating you with some useful info.
The difficulties I had with continuation runs in Chimera took several weeks to resolve, but now I can get to ~200ps as opposed to the 5ps value of a standard run.
The problem lies in the Prep Structure interface (attached). The instruction "Input restart-trajectory file from previous equilibration/production" would reasonably be interpreted as inputting the restart-trajectory file from an earlier run? This file is generated after each run and is posted on the desktop. However, it's never accepted, and the error message I mentioned in an earlier email pops up every time.
It actually needs to have a suffix <01. Which is very odd,as such a file doesn't actually exist until the run is over! Adding a suffixes of unit value higher to the output files is however logical.
It might be just ambiguous interface design, something that slipped thru the net, but the problem is very simply resolved by this method. If only it hadn't taken me weeks to stumble on it!
Still very happy with Chimera, getting excellent data for my dissertation. Best wishes Simon
On 17 January 2018 at 18:38, Elaine Meng <meng@cgl.ucsf.edu <mailto:meng@cgl.ucsf.edu>> wrote: As I understand it, the message is that the molecules in the input restart file are not the same as the current molecules (not exactly the same set of residues, atoms, bonds). Changing the filenames would allow for a completely separate run, but it would not solve the problem of continuing the same simulation if the chemical system is different. How it might be different, I don’t know. All I can say is if you changed something between the runs, don’t do that. Elaine
On Jan 17, 2018, at 4:33 AM, simon chapman <rowanlodge19@gmail.com <mailto:rowanlodge19@gmail.com>> wrote:
Hello Eric and Elaine. Chance of earning a second pint of ginger beer if you wish...
Simulations and analysis going very well, I have substituted all 12 oxygens for sulphur. Each requires a new name: UNK I, UNK2 etc. Advice for others...naming will only alow digits 1 to 9. so for 12 iterations, I used zero and the original unappended "UNK". Which makes 12. I have also successfully set production for 50000steps, equivalent to 8ps.
However, there appears to be an option for continuation from one run to a subsequent one. I've tried for a few days now, but get the same error message every time ('continuation2 attachment'). Changing file names as described in the message doesn't work. It's probably something simple (probably me in fact), but would really appreciate your comments.
Best wishes Simon
On 10 January 2018 at 19:01, Eric Pettersen <pett@cgl.ucsf.edu <mailto:pett@cgl.ucsf.edu>> wrote: Great! If we’re in the neighborhood I’ll take a pint of ginger ale. :-)
—Eric
On Jan 10, 2018, at 4:09 AM, simon chapman <rowanlodge19@gmail.com <mailto:rowanlodge19@gmail.com>> wrote:
Hello Elaine and Eric. Thankyou for the prompt reply....and I'm very impressed! Eric's suggestion #2 worked!
I assumed the protonation was OK as I ran the PDB downloaded structure through a protonation webserver (H++). So I just changed the name of the novel substitution. It looks like additional alterations will also need renaming: I ran the next substitution in the incremental progression( #5) as "UNK3",and that is just about to complete.
I am especially pleased as the data are definitely tending to support my hypothesis. You will acknowledged in the Methodology section of my dissertation.
So, thanks again. There's a charming 14th century pub just down the lane from me...buy you both a pint next time you're passing through
Best wishes Simon
On 9 January 2018 at 19:31, Eric Pettersen <pett@cgl.ucsf.edu <mailto:pett@cgl.ucsf.edu>> wrote: Hi Simon, Everything Elaine says is true. I do have some last ditch suggestions for you though. One thing is that the fourth guanine differs in protonation state from the others in that the fourth one either has or lacks an HO3’ whereas the others don’t. You should investigate why this is the case. The two possibilities are then:
1) The protonation difference is an error. You would need to correct the structure so that all four guanines start with the same protonation. 2) For some reason the protonation difference is okay. You then need to give the modified version of the fourth guanine a different name from the others, e.g. “UK2” instead of “UNK”. Not guaranteeing this will work, but it might.
—Eric
Eric Pettersen UCSF Computer Graphics Lab
On Jan 9, 2018, at 10:24 AM, Elaine Meng <meng@cgl.ucsf.edu <mailto:meng@cgl.ucsf.edu>> wrote:
Hi Simon, I think you may be stretching the tools beyond their intended purposes: Chimera’s minimization, molecular dynamics interface, and automatic parametrization of nonstandard residues with Antechamber are meant to provide relatively simple access to such calculations by noncomputational scientists, given the large learning curves of dedicated simulation packages like GROMACS, AMBER, etc. These tools can be highly useful to clean up structures or suggest additional reasonable conformations. However, they are not meant for precise quantification of energies or for very long simulations, especially not in conjunction with nonstandard residues with parameters estimated by Antechamber. The Gasteiger charges are very quick-and-dirty, so I would caution against overinterpreting simulations that employ them. Further, Antechember is meant to cover most small organic molecules but not every possible molecule.
It seems that the modified UNK residues fall into two structural types (as per the “specify net charges” dialog), and that one (maybe just the 4th?) fails in parametrization by Antechamber, and then the Solvate or Add Ions tool cannot find some parameter needed for its calculation. These tools come from the Ambertools package even though they are included with Chimera and Chimera has interfaces to them. I don’t know exactly what the problem is, nor do I have a solution, sorry. Elaine ----- Elaine C. Meng, Ph.D. UCSF Chimera(X) team Department of Pharmaceutical Chemistry University of California, San Francisco
On Jan 9, 2018, at 1:30 AM, simon chapman <rowanlodge19@gmail.com <mailto:rowanlodge19@gmail.com>> wrote:
Hello Elaine, sorry to bother you again.
Chimera has been a real treat so far, but I have hit a problem. I've struggled for a week now, and finally given up!
I am incrementally substituting S for O on four guanines in a regular complex. 3 substitutions work fine in a simulation and producing very encouraging data (attachment 1) But 4 substitutions (attachment 2) triggers a load of error messages. The first is attachment 3. After solvation, attachment 4 appears and the solvation box is removed if neutralising ions are added. If ions not added, attachment 5 warning appears. Not sure if attachment 6 is relevant.
Hope you can help/advise?
Best wishes Simon
<prod phase for chimera team.png>_______________________________________________ Chimera-users mailing list: Chimera-users@cgl.ucsf.edu <mailto:Chimera-users@cgl.ucsf.edu> Manage subscription: http://plato.cgl.ucsf.edu/mailman/listinfo/chimera-users <http://plato.cgl.ucsf.edu/mailman/listinfo/chimera-users>
participants (3)
-
 Elaine Meng
Elaine Meng -
 Eric Pettersen
Eric Pettersen -
 simon chapman
simon chapman