
Hi, I'm very impressed with Chimera as a user of only 3 weeks so far Trying out the various facilities has been a very useful exercise for completing my Masters in MedChem. However, when I emulated the tutorial covering COX 1 and 2 inhibition, inputting the molecules fluribrofen and SC558 I generated via DS Visualiser bind at the outside edge of the enzymes. Not centrally as displayed in the video. I also noted the molecular data in ViewDock, ie: energies etc, is missing. The table is blank other than the molecule number. Selecting 'Column' etc has no effect. The 'H-bonds' option does display however. I've probably done something wrong, hey ho...but can you enlighten me? Best wishes Simon

HI Simon, We’re glad Chimera has been helpful in your studies! The COX inhibitors demo (under Tools… Demos in the menu) just uses some structures from RCSB PDB that already have the small molecules in the correct locations relative to the protein structures. It’s not really a tutorial (it doesn’t tell you how to do anything) but a series of actions in Chimera that show you parts of these existing structures. The Credits panel of that demo says which PDB entries were used: 1cqe (COX-1 with flurbiprofen) and 6cox (COX-2 with SC-558). So if you just start Chimera and use command “open 1cqe” for example, it will show the COX-1 complex structure. 1cqe and 6cox each contain a homodimer (two copies of the protein+ligand), but for simplicity in the demos, only monomers were shown. I don’t know where you got the structures you opened, so the coordinates might be completely different. I.e., maybe what you see is where DSV put the small molecules. Also ViewDock will only display energies if what you read in to ViewDock is (1) a format that VIewDock knows, and (2) actually includes those energies. Chimera doesn’t calculate them for you. I have no idea what you opened in ViewDock. Its manual page lists the types of docking outputs that it knows how to read, like from the programs DOCK, Glide, AutoDock, GOLD, etc. <http://www.rbvi.ucsf.edu/chimera/docs/ContributedSoftware/viewdock/framevd.h...> There is a ViewDock tutorial that includes sample input from DOCK: <http://www.rbvi.ucsf.edu/chimera/docs/UsersGuide/tutorials/vdtut.html> If you meant you tried the Autodock Vina interface in Chimera and it didn’t put the small molecules in the right place, that is the way of docking calculations. There are a lot of docking parameters and setup options and sometimes adjusting those will get the right answer, or simply sampling more orientations. Unfortunately the interface in Chimera uses a web service that doesn’t allow very much sampling. To do a thorough docking study, you might have to obtain a docking program and run it separately from Chimera. However, no need to do docking for these particular molecules because the structures of the complexes are already known and publicly available from the PDB! I hope this helps, Elaine ----- Elaine C. Meng, Ph.D. UCSF Chimera(X) team Department of Pharmaceutical Chemistry University of California, San Francisco
On Sep 6, 2017, at 4:17 AM, simon chapman <rowanlodge19@gmail.com> wrote:
Hi, I'm very impressed with Chimera as a user of only 3 weeks so far
Trying out the various facilities has been a very useful exercise for completing my Masters in MedChem.
However, when I emulated the tutorial covering COX 1 and 2 inhibition, inputting the molecules fluribrofen and SC558 I generated via DS Visualiser bind at the outside edge of the enzymes. Not centrally as displayed in the video.
I also noted the molecular data in ViewDock, ie: energies etc, is missing. The table is blank other than the molecule number. Selecting 'Column' etc has no effect. The 'H-bonds' option does display however.
I've probably done something wrong, hey ho...but can you enlighten me?
Best wishes Simon
Very kind Elaine. Thank you. I'll follow up your comments. Hope I don't have to bother you too much again, but there will probably be occasions when I hit a brick wall... It's all a part of the learning process!! Best wishes Simon On 6 September 2017 at 17:59, Elaine Meng <meng@cgl.ucsf.edu> wrote:
HI Simon, We’re glad Chimera has been helpful in your studies!
The COX inhibitors demo (under Tools… Demos in the menu) just uses some structures from RCSB PDB that already have the small molecules in the correct locations relative to the protein structures. It’s not really a tutorial (it doesn’t tell you how to do anything) but a series of actions in Chimera that show you parts of these existing structures. The Credits panel of that demo says which PDB entries were used: 1cqe (COX-1 with flurbiprofen) and 6cox (COX-2 with SC-558). So if you just start Chimera and use command “open 1cqe” for example, it will show the COX-1 complex structure. 1cqe and 6cox each contain a homodimer (two copies of the protein+ligand), but for simplicity in the demos, only monomers were shown.
I don’t know where you got the structures you opened, so the coordinates might be completely different. I.e., maybe what you see is where DSV put the small molecules. Also ViewDock will only display energies if what you read in to ViewDock is (1) a format that VIewDock knows, and (2) actually includes those energies. Chimera doesn’t calculate them for you. I have no idea what you opened in ViewDock. Its manual page lists the types of docking outputs that it knows how to read, like from the programs DOCK, Glide, AutoDock, GOLD, etc. <http://www.rbvi.ucsf.edu/chimera/docs/ContributedSoftware/viewdock/ framevd.html>
There is a ViewDock tutorial that includes sample input from DOCK: <http://www.rbvi.ucsf.edu/chimera/docs/UsersGuide/tutorials/vdtut.html>
If you meant you tried the Autodock Vina interface in Chimera and it didn’t put the small molecules in the right place, that is the way of docking calculations. There are a lot of docking parameters and setup options and sometimes adjusting those will get the right answer, or simply sampling more orientations. Unfortunately the interface in Chimera uses a web service that doesn’t allow very much sampling. To do a thorough docking study, you might have to obtain a docking program and run it separately from Chimera.
However, no need to do docking for these particular molecules because the structures of the complexes are already known and publicly available from the PDB!
I hope this helps, Elaine ----- Elaine C. Meng, Ph.D. UCSF Chimera(X) team Department of Pharmaceutical Chemistry University of California, San Francisco
On Sep 6, 2017, at 4:17 AM, simon chapman <rowanlodge19@gmail.com> wrote:
Hi, I'm very impressed with Chimera as a user of only 3 weeks so far
Trying out the various facilities has been a very useful exercise for completing my Masters in MedChem.
However, when I emulated the tutorial covering COX 1 and 2 inhibition, inputting the molecules fluribrofen and SC558 I generated via DS Visualiser bind at the outside edge of the enzymes. Not centrally as displayed in the video.
I also noted the molecular data in ViewDock, ie: energies etc, is missing. The table is blank other than the molecule number. Selecting 'Column' etc has no effect. The 'H-bonds' option does display however.
I've probably done something wrong, hey ho...but can you enlighten me?
Best wishes Simon
Hello again Elaine. I understand now about the COX demo... I used DSV and PDB to generate the structures. Obviously had incorrect co-ordinates. Will watch out for that next time. However, following the tutorial exactly for ViewDock still doesn't quite match. Attached screenshot shows tutorial pic and the version that I get... the onscreen background molecular structure is identical with the 'official' one. So, not sure why the energy data isn't showing up?? Also, is there a way to slow down rotation of a molecule after command 'roll'? I notice your name is at the top of the data slide...is the video your production? Best wishes Simon On 6 September 2017 at 17:59, Elaine Meng <meng@cgl.ucsf.edu> wrote:
HI Simon, We’re glad Chimera has been helpful in your studies!
The COX inhibitors demo (under Tools… Demos in the menu) just uses some structures from RCSB PDB that already have the small molecules in the correct locations relative to the protein structures. It’s not really a tutorial (it doesn’t tell you how to do anything) but a series of actions in Chimera that show you parts of these existing structures. The Credits panel of that demo says which PDB entries were used: 1cqe (COX-1 with flurbiprofen) and 6cox (COX-2 with SC-558). So if you just start Chimera and use command “open 1cqe” for example, it will show the COX-1 complex structure. 1cqe and 6cox each contain a homodimer (two copies of the protein+ligand), but for simplicity in the demos, only monomers were shown.
I don’t know where you got the structures you opened, so the coordinates might be completely different. I.e., maybe what you see is where DSV put the small molecules. Also ViewDock will only display energies if what you read in to ViewDock is (1) a format that VIewDock knows, and (2) actually includes those energies. Chimera doesn’t calculate them for you. I have no idea what you opened in ViewDock. Its manual page lists the types of docking outputs that it knows how to read, like from the programs DOCK, Glide, AutoDock, GOLD, etc. <http://www.rbvi.ucsf.edu/chimera/docs/ContributedSoftware/viewdock/ framevd.html>
There is a ViewDock tutorial that includes sample input from DOCK: <http://www.rbvi.ucsf.edu/chimera/docs/UsersGuide/tutorials/vdtut.html>
If you meant you tried the Autodock Vina interface in Chimera and it didn’t put the small molecules in the right place, that is the way of docking calculations. There are a lot of docking parameters and setup options and sometimes adjusting those will get the right answer, or simply sampling more orientations. Unfortunately the interface in Chimera uses a web service that doesn’t allow very much sampling. To do a thorough docking study, you might have to obtain a docking program and run it separately from Chimera.
However, no need to do docking for these particular molecules because the structures of the complexes are already known and publicly available from the PDB!
I hope this helps, Elaine ----- Elaine C. Meng, Ph.D. UCSF Chimera(X) team Department of Pharmaceutical Chemistry University of California, San Francisco
On Sep 6, 2017, at 4:17 AM, simon chapman <rowanlodge19@gmail.com> wrote:
Hi, I'm very impressed with Chimera as a user of only 3 weeks so far
Trying out the various facilities has been a very useful exercise for completing my Masters in MedChem.
However, when I emulated the tutorial covering COX 1 and 2 inhibition, inputting the molecules fluribrofen and SC558 I generated via DS Visualiser bind at the outside edge of the enzymes. Not centrally as displayed in the video.
I also noted the molecular data in ViewDock, ie: energies etc, is missing. The table is blank other than the molecule number. Selecting 'Column' etc has no effect. The 'H-bonds' option does display however.
I've probably done something wrong, hey ho...but can you enlighten me?
Best wishes Simon
{kind=link}
Hi Simon, You missed a step or two in the ViewDock tutorial. You have to use the Columns menu to control what the dialog is showing. See the paragraph in the tutorial starting “The docked compounds are enumerated…” <http://www.rbvi.ucsf.edu/chimera/docs/UsersGuide/tutorials/vdtut.html> There are several options to the “roll” command including number of degrees per frame. For a slower roll, you can set this to a low number, e.g. “roll y 0.1”. Use command “help roll” to see the manual page, or view the copy at our website: <http://www.rbvi.ucsf.edu/chimera/docs/UsersGuide/midas/roll.html> … it says the default is 1.5 degrees per frame. If you already started a roll with inifinite frames, you can use “freeze” to halt it. <http://www.rbvi.ucsf.edu/chimera/docs/UsersGuide/midas/freeze.html> If you mean the COX demo credits, yes, I created that demo. It is really a series of Chimera commands to execute for each “panel." Users can create their own demos, but it requires some familiarity with the commands. <http://www.rbvi.ucsf.edu/chimera/docs/ContributedSoftware/demos/demos.html> Best, Elaine ----- Elaine C. Meng, Ph.D. UCSF Chimera(X) team Department of Pharmaceutical Chemistry University of California, San Francisco
On Sep 8, 2017, at 6:57 AM, simon chapman <rowanlodge19@gmail.com> wrote:
Hello again Elaine. I understand now about the COX demo... I used DSV and PDB to generate the structures. Obviously had incorrect co-ordinates. Will watch out for that next time.
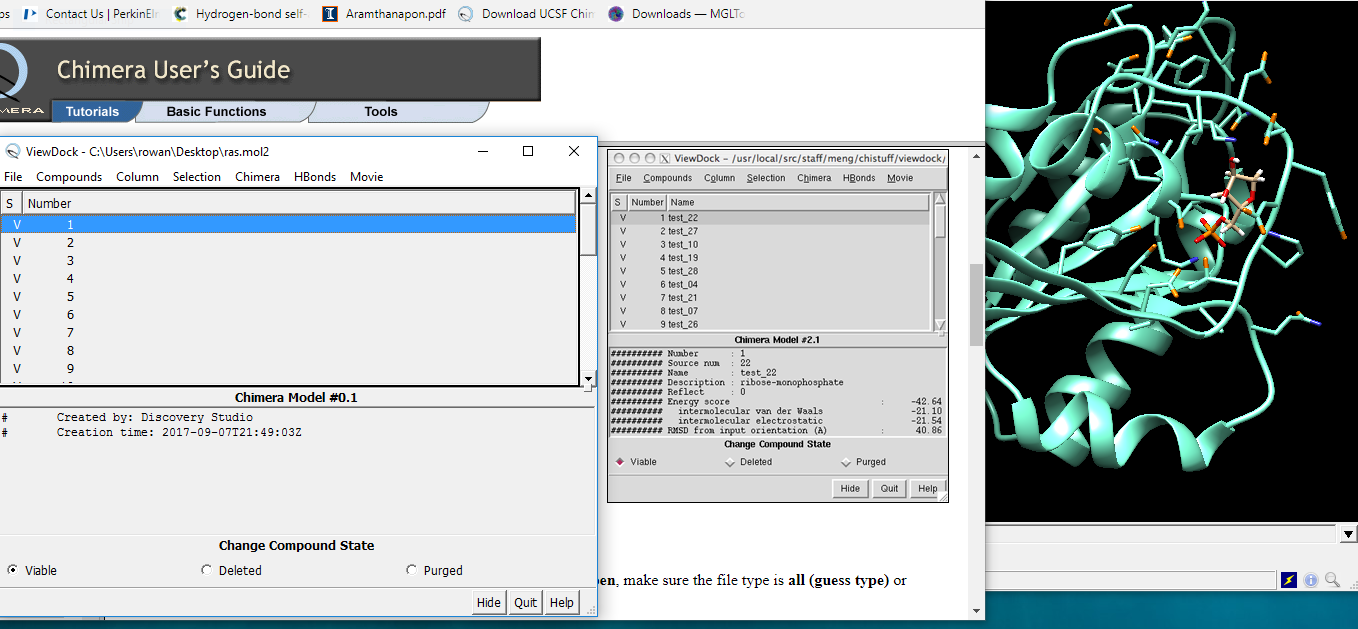
However, following the tutorial exactly for ViewDock still doesn't quite match. Attached screenshot shows tutorial pic and the version that I get... the onscreen background molecular structure is identical with the 'official' one. So, not sure why the energy data isn't showing up??
Also, is there a way to slow down rotation of a molecule after command 'roll'?
I notice your name is at the top of the data slide...is the video your production?
Best wishes Simon
Hello Elaine, apologies for another email..! I selected 'Columns' on the first occasion I followed the tutorial. It only selects Show, Hide ,Read and Display. clicking the first two just shows 'Number' . Selecting that does nothing at all. 'Display' shows 2D structures as expected. I get the same result every time I've tried since. I've left- and right-clicked all the options several times,but it seems the energy data simply isn't available... Got the rotate speed sorted immediately along all 3 axes, so thanks for that. I have also made a (shaky) demo from the instruction link you provided. Best wishes Simon On 8 September 2017 at 17:16, Elaine Meng <meng@cgl.ucsf.edu> wrote:
Hi Simon, You missed a step or two in the ViewDock tutorial. You have to use the Columns menu to control what the dialog is showing. See the paragraph in the tutorial starting “The docked compounds are enumerated…” <http://www.rbvi.ucsf.edu/chimera/docs/UsersGuide/tutorials/vdtut.html>
There are several options to the “roll” command including number of degrees per frame. For a slower roll, you can set this to a low number, e.g. “roll y 0.1”. Use command “help roll” to see the manual page, or view the copy at our website: <http://www.rbvi.ucsf.edu/chimera/docs/UsersGuide/midas/roll.html> … it says the default is 1.5 degrees per frame.
If you already started a roll with inifinite frames, you can use “freeze” to halt it. <http://www.rbvi.ucsf.edu/chimera/docs/UsersGuide/midas/freeze.html>
If you mean the COX demo credits, yes, I created that demo. It is really a series of Chimera commands to execute for each “panel." Users can create their own demos, but it requires some familiarity with the commands. <http://www.rbvi.ucsf.edu/chimera/docs/ContributedSoftware/demos/ demos.html>
Best, Elaine ----- Elaine C. Meng, Ph.D. UCSF Chimera(X) team Department of Pharmaceutical Chemistry University of California, San Francisco
On Sep 8, 2017, at 6:57 AM, simon chapman <rowanlodge19@gmail.com> wrote:
Hello again Elaine. I understand now about the COX demo... I used DSV and PDB to generate the structures. Obviously had incorrect co-ordinates. Will watch out for that next time.
However, following the tutorial exactly for ViewDock still doesn't quite match. Attached screenshot shows tutorial pic and the version that I get... the onscreen background molecular structure is identical with the 'official' one. So, not sure why the energy data isn't showing up??
Also, is there a way to slow down rotation of a molecule after command 'roll'?
I notice your name is at the top of the data slide...is the video your production?
Best wishes Simon
Hi Simon, I see in the screenshot image you sent in the earlier e-mail (yesterday) that your ViewDock dialog with the “ras.mol2” data opened in it says “Created by: Discovery Studio”. Thus I can only guess that you’d saved the “ras.mol2” file from that program, and that it either removed or reformatted the energy information so that Chimera could not read it. Try downloading the ras.mol2 file again from the link in the tutorial, saving it as plain text and using it in Chimera WITHOUT opening it in DIscovery Studio first and it should work. Kudos on making your own demo! By the way, all of the User Guide is included with your download, and you can search it from the Help menu. Best, Elaine
On Sep 9, 2017, at 6:42 AM, simon chapman <rowanlodge19@gmail.com> wrote:
Hello Elaine, apologies for another email..!
I selected 'Columns' on the first occasion I followed the tutorial. It only selects Show, Hide ,Read and Display. clicking the first two just shows 'Number' . Selecting that does nothing at all. 'Display' shows 2D structures as expected. I get the same result every time I've tried since. I've left- and right-clicked all the options several times,but it seems the energy data simply isn't available...
Got the rotate speed sorted immediately along all 3 axes, so thanks for that.
I have also made a (shaky) demo from the instruction link you provided.
Best wishes Simon
On 8 September 2017 at 17:16, Elaine Meng <meng@cgl.ucsf.edu> wrote: Hi Simon, You missed a step or two in the ViewDock tutorial. You have to use the Columns menu to control what the dialog is showing. See the paragraph in the tutorial starting “The docked compounds are enumerated…” <http://www.rbvi.ucsf.edu/chimera/docs/UsersGuide/tutorials/vdtut.html>
There are several options to the “roll” command including number of degrees per frame. For a slower roll, you can set this to a low number, e.g. “roll y 0.1”. Use command “help roll” to see the manual page, or view the copy at our website: <http://www.rbvi.ucsf.edu/chimera/docs/UsersGuide/midas/roll.html> … it says the default is 1.5 degrees per frame.
If you already started a roll with inifinite frames, you can use “freeze” to halt it. <http://www.rbvi.ucsf.edu/chimera/docs/UsersGuide/midas/freeze.html>
If you mean the COX demo credits, yes, I created that demo. It is really a series of Chimera commands to execute for each “panel." Users can create their own demos, but it requires some familiarity with the commands. <http://www.rbvi.ucsf.edu/chimera/docs/ContributedSoftware/demos/demos.html>
Best, Elaine ----- Elaine C. Meng, Ph.D. UCSF Chimera(X) team Department of Pharmaceutical Chemistry University of California, San Francisco
On Sep 8, 2017, at 6:57 AM, simon chapman <rowanlodge19@gmail.com> wrote:
Hello again Elaine. I understand now about the COX demo... I used DSV and PDB to generate the structures. Obviously had incorrect co-ordinates. Will watch out for that next time.
However, following the tutorial exactly for ViewDock still doesn't quite match. Attached screenshot shows tutorial pic and the version that I get... the onscreen background molecular structure is identical with the 'official' one. So, not sure why the energy data isn't showing up??
Also, is there a way to slow down rotation of a molecule after command 'roll'?
I notice your name is at the top of the data slide...is the video your production?
Best wishes Simon
_______________________________________________ Chimera-users mailing list: Chimera-users@cgl.ucsf.edu Manage subscription: http://plato.cgl.ucsf.edu/mailman/listinfo/chimera-users
Thank you Elaine...that did it! Couldn't save as Plaintext, but it still worked as Notepad. I am very pleased with the MD Movie tutorial...it worked for me without too many changes, and is suitably impressive. Did the authors supplying the data derive them from AMBER? How can I supply data relevant to the molecules I'm studying to produce the appropriate MD Movie? Also, and I hope it's OK to bother you again, the attached screenshots show the result of trying to download 1plx.pdb. I've tried several formats, but the message "no pdb structures found" appears every time. Clicking " 1plx.pdb" in the tutorial leads to the page shown. Selecting "save as..." leads to the other two pop-ups in screenshot 1. Downloads lists the file as Tutorial FrameSet, and contains the four files shown in screenshot 2. Feel like I'm going round in circles with this one. Thanks in advance...despite the challenges, Chimera is very impressive, and should enhance my MSc thesis which commences next month Best wishes Simon On 10 September 2017 at 01:49, Elaine Meng <meng@cgl.ucsf.edu> wrote:
Hi Simon, I see in the screenshot image you sent in the earlier e-mail (yesterday) that your ViewDock dialog with the “ras.mol2” data opened in it says “Created by: Discovery Studio”. Thus I can only guess that you’d saved the “ras.mol2” file from that program, and that it either removed or reformatted the energy information so that Chimera could not read it. Try downloading the ras.mol2 file again from the link in the tutorial, saving it as plain text and using it in Chimera WITHOUT opening it in DIscovery Studio first and it should work.
Kudos on making your own demo! By the way, all of the User Guide is included with your download, and you can search it from the Help menu. Best, Elaine
On Sep 9, 2017, at 6:42 AM, simon chapman <rowanlodge19@gmail.com> wrote:
Hello Elaine, apologies for another email..!
I selected 'Columns' on the first occasion I followed the tutorial. It only selects Show, Hide ,Read and Display. clicking the first two just shows 'Number' . Selecting that does nothing at all. 'Display' shows 2D structures as expected. I get the same result every time I've tried since. I've left- and right-clicked all the options several times,but it seems the energy data simply isn't available...
Got the rotate speed sorted immediately along all 3 axes, so thanks for that.
I have also made a (shaky) demo from the instruction link you provided.
Best wishes Simon
On 8 September 2017 at 17:16, Elaine Meng <meng@cgl.ucsf.edu> wrote: Hi Simon, You missed a step or two in the ViewDock tutorial. You have to use the Columns menu to control what the dialog is showing. See the paragraph in the tutorial starting “The docked compounds are enumerated…” <http://www.rbvi.ucsf.edu/chimera/docs/UsersGuide/tutorials/vdtut.html>
There are several options to the “roll” command including number of degrees per frame. For a slower roll, you can set this to a low number, e.g. “roll y 0.1”. Use command “help roll” to see the manual page, or view the copy at our website: <http://www.rbvi.ucsf.edu/chimera/docs/UsersGuide/midas/roll.html> … it says the default is 1.5 degrees per frame.
If you already started a roll with inifinite frames, you can use “freeze” to halt it. <http://www.rbvi.ucsf.edu/chimera/docs/UsersGuide/midas/freeze.html>
If you mean the COX demo credits, yes, I created that demo. It is really a series of Chimera commands to execute for each “panel." Users can create their own demos, but it requires some familiarity with the commands. <http://www.rbvi.ucsf.edu/chimera/docs/ContributedSoftware/demos/ demos.html>
Best, Elaine ----- Elaine C. Meng, Ph.D. UCSF Chimera(X) team Department of Pharmaceutical Chemistry University of California, San Francisco
On Sep 8, 2017, at 6:57 AM, simon chapman <rowanlodge19@gmail.com> wrote:
Hello again Elaine. I understand now about the COX demo... I used DSV and PDB to generate the structures. Obviously had incorrect co-ordinates. Will watch out for that next time.
However, following the tutorial exactly for ViewDock still doesn't quite match. Attached screenshot shows tutorial pic and the version that I get... the onscreen background molecular structure is identical with the 'official' one. So, not sure why the energy data isn't showing up??
Also, is there a way to slow down rotation of a molecule after command 'roll'?
I notice your name is at the top of the data slide...is the video your production?
Best wishes Simon
_______________________________________________ Chimera-users mailing list: Chimera-users@cgl.ucsf.edu Manage subscription: http://plato.cgl.ucsf.edu/ mailman/listinfo/chimera-users
{kind=link}
{kind=link}
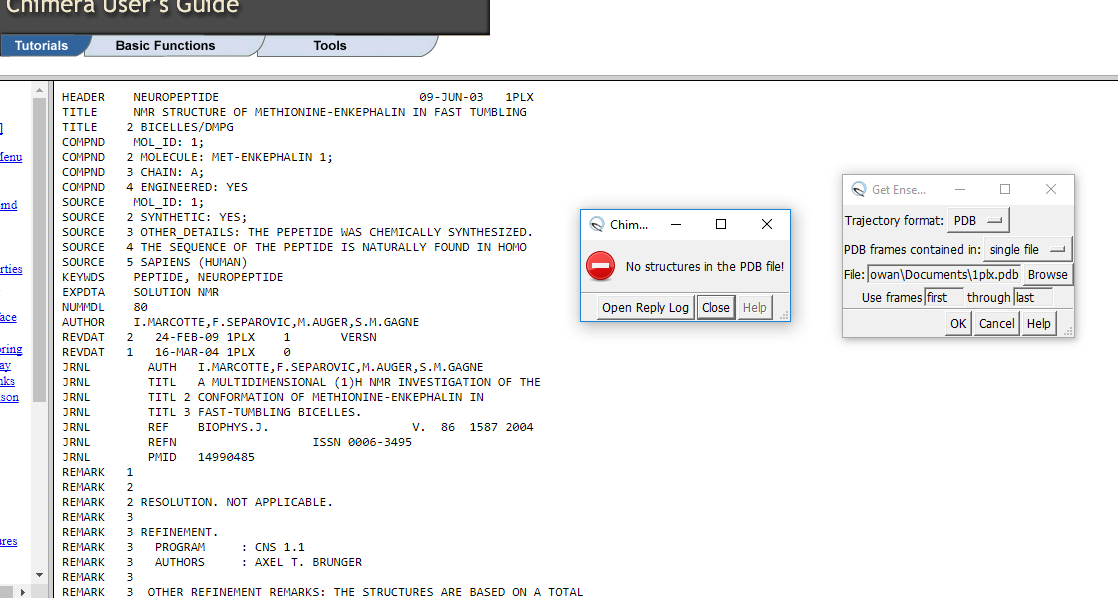
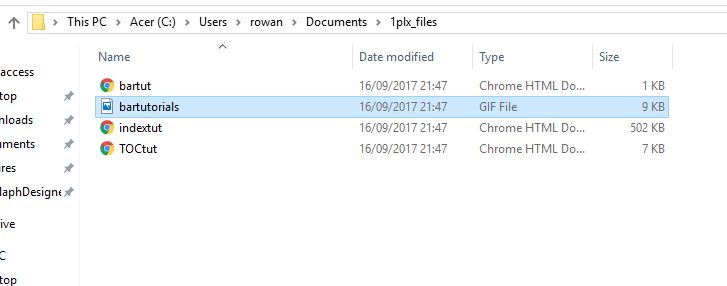
Hi Simon, (1) There are lots of molecular dynamics programs: AMBER, GROMACS, NAMD etc., and the list of input formats for the Chimera “MD Movie” tool shows which of those programs’ results it can handle: <http://www.rbvi.ucsf.edu/chimera/docs/ContributedSoftware/movie/framemovie.h...> Chimera itself even has a Molecular Dynamics SImulation tool (see Tools… MD/Ensemble Analysis) but it is relatively slow. Be aware that there is a learning curve to running MD simulations. Garbage in gives garbage out, so you have to take some time to learn about the technique and its limitations. There are many input options and parameters, and just taking all the defaults isn’t usually the path to success for any given molecular system. There are textbooks on the subject and running a decent simulation could be a project in itself. Or if you just wanted a conformational ensemble rather than a trajectory, there are a whole host of other programs that do some normal mode analysis based on network models. Those seem simpler than MD to use, but the results are more limited; that’s just a general impression… I’m not an expert on the topic. Even simpler is if you have two different conformations of some protein, you can morph between them in Chimera and show the results in Chimera. For example, see the “superpositions and alignments” tutorial starting at “different conformations of the same protein" <http://www.rbvi.ucsf.edu/chimera/docs/UsersGuide/tutorials/alignments.html> (2) References to the papers about the data are given in the MD Movie tutorial. Part 1 uses MD results (probably from AMBER but you’d have to read the paper to doublecheck) and part 2 uses an NMR ensemble. <http://www.rbvi.ucsf.edu/chimera/docs/UsersGuide/tutorials/ensembles2.html> (3) the browser is showing the text of 1plx.pdb … you just need to save it as plain text, just like you did for ras.mol2 . Alternatively, you can get the same file from RCSB PDB, Download Files -> PDB Format: <http://www.rcsb.org/pdb/explore/explore.do?structureId=1plx> I hope this helps, Elaine ----- Elaine C. Meng, Ph.D. UCSF Chimera(X) team Department of Pharmaceutical Chemistry University of California, San Francisco
On Sep 16, 2017, at 2:04 PM, simon chapman <rowanlodge19@gmail.com> wrote:
Thank you Elaine...that did it! Couldn't save as Plaintext, but it still worked as Notepad.
I am very pleased with the MD Movie tutorial...it worked for me without too many changes, and is suitably impressive. Did the authors supplying the data derive them from AMBER? How can I supply data relevant to the molecules I'm studying to produce the appropriate MD Movie?
Also, and I hope it's OK to bother you again, the attached screenshots show the result of trying to download 1plx.pdb. I've tried several formats, but the message "no pdb structures found" appears every time. Clicking " 1plx.pdb" in the tutorial leads to the page shown. Selecting "save as..." leads to the other two pop-ups in screenshot 1. Downloads lists the file as Tutorial FrameSet, and contains the four files shown in screenshot 2. Feel like I'm going round in circles with this one.
Thanks in advance...despite the challenges, Chimera is very impressive, and should enhance my MSc thesis which commences next month
Best wishes Simon

Is there a way to keep & show the ligands in the Morph between structures?. In the morphs that I've tried the ligands do not show up. Thanks H.
Sorry that was an old question and here was the answer. ---------------------------------------------------------- As I had understood it (and had written in the documentation) <http://www.rbvi.ucsf.edu/chimera/docs/ContributedSoftware/morph/morph.html#p...>: "HET residues such as ligands and ions are only included if they are present in both structures and attached to the same atom(s) in the paired chains by at least one "covalent" bond (which can be an unrealistic bond added manually, e.g., with the command bond, and subsequently undisplayed) or ion coordination pseudobond. Once residues are paired, atoms in common within those residues are paired. In paired residues of the same type, atom pairing is straightforward. In paired residues of different types, only atoms with the same names are paired, and only a single connected fragment is kept per residue." I hope this helps, Elaine ---------- Elaine C. Meng, Ph.D. UCSF Chimera(X) team Department of Pharmaceutical Chemistry University of California, San Francisco -----Original Message----- From: Chimera-users [mailto:chimera-users-bounces@cgl.ucsf.edu] On Behalf Of Hernando J Sosa Sent: Sunday, September 17, 2017 8:00 PM To: chimera List Subject: [Chimera-users] morph & ligands [This sender failed our fraud detection checks and may not be who they appear to be. Learn about spoofing at http://aka.ms/LearnAboutSpoofing] Is there a way to keep & show the ligands in the Morph between structures?. In the morphs that I've tried the ligands do not show up. Thanks H. _______________________________________________ Chimera-users mailing list: Chimera-users@cgl.ucsf.edu Manage subscription: http://plato.cgl.ucsf.edu/mailman/listinfo/chimera-users
Hi Hernando, While the morph (trajectory) model itself may not include the ligand, it is very common to simply display the ligand(s) from the other, input models. You would just make sure that the model with the ligand of interest is shown (e.g. with “Shown” checkbox in Model Panel), and then hide all of its other atoms and ribbons that you don’t want to show. Even fancier, with a little more work you could (A) make the ligand appear only at a certain point in the morph playback instead of the whole time, or (B) make it fly into the binding site. There is an example of (A) in the animation gallery. See the first movie, “kinase morph” and its command script. The “coordset” command is used to play back the previously calculated morph trajectory model #3. There are several “coordset” commands to play back different parts of the trajectory, interspersed with commands to show the ligand residue ACP, some sidechains, and 2D labels. <http://www.rbvi.ucsf.edu/chimera/animations/animations.html#kinase-morph> There is an example of (B) in this movie-making tutorial: <http://www.rbvi.ucsf.edu/chimera/data/tutorials/movies08/moviemaking.html> I hope this helps, Elaine ---------- Elaine C. Meng, Ph.D. UCSF Chimera(X) team Department of Pharmaceutical Chemistry University of California, San Francisco
On Sep 18, 2017, at 8:30 AM, Hernando J Sosa <hernando.sosa@einstein.yu.edu> wrote:
Sorry that was an old question and here was the answer.
---------------------------------------------------------- As I had understood it (and had written in the documentation) <http://www.rbvi.ucsf.edu/chimera/docs/ContributedSoftware/morph/morph.html#p...>:
"HET residues such as ligands and ions are only included if they are present in both structures and attached to the same atom(s) in the paired chains by at least one "covalent" bond (which can be an unrealistic bond added manually, e.g., with the command bond, and subsequently undisplayed) or ion coordination pseudobond.
Once residues are paired, atoms in common within those residues are paired. In paired residues of the same type, atom pairing is straightforward. In paired residues of different types, only atoms with the same names are paired, and only a single connected fragment is kept per residue."
I hope this helps, Elaine ---------- Elaine C. Meng, Ph.D. UCSF Chimera(X) team Department of Pharmaceutical Chemistry University of California, San Francisco
-----Original Message----- From: Chimera-users [mailto:chimera-users-bounces@cgl.ucsf.edu] On Behalf Of Hernando J Sosa Sent: Sunday, September 17, 2017 8:00 PM To: chimera List Subject: [Chimera-users] morph & ligands
[This sender failed our fraud detection checks and may not be who they appear to be. Learn about spoofing at http://aka.ms/LearnAboutSpoofing]
Is there a way to keep & show the ligands in the Morph between structures?. In the morphs that I've tried the ligands do not show up.
Thanks
H.
Thanks Elaine. I did try making a covalent bond with the bond command between the protein and the ligand (same atoms bonded in the two conformers) but that didn't work; the ligand still disappear in the morph movie. I'll try the examples in the tutorial. Best H. -----Original Message----- From: Elaine Meng [mailto:meng@cgl.ucsf.edu] Sent: Monday, September 18, 2017 12:17 PM To: Hernando J Sosa Cc: chimera List Subject: Re: [Chimera-users] morph & ligands Hi Hernando, While the morph (trajectory) model itself may not include the ligand, it is very common to simply display the ligand(s) from the other, input models. You would just make sure that the model with the ligand of interest is shown (e.g. with “Shown” checkbox in Model Panel), and then hide all of its other atoms and ribbons that you don’t want to show. Even fancier, with a little more work you could (A) make the ligand appear only at a certain point in the morph playback instead of the whole time, or (B) make it fly into the binding site. There is an example of (A) in the animation gallery. See the first movie, “kinase morph” and its command script. The “coordset” command is used to play back the previously calculated morph trajectory model #3. There are several “coordset” commands to play back different parts of the trajectory, interspersed with commands to show the ligand residue ACP, some sidechains, and 2D labels. <http://www.rbvi.ucsf.edu/chimera/animations/animations.html#kinase-morph> There is an example of (B) in this movie-making tutorial: <http://www.rbvi.ucsf.edu/chimera/data/tutorials/movies08/moviemaking.html> I hope this helps, Elaine ---------- Elaine C. Meng, Ph.D. UCSF Chimera(X) team Department of Pharmaceutical Chemistry University of California, San Francisco
On Sep 18, 2017, at 8:30 AM, Hernando J Sosa <hernando.sosa@einstein.yu.edu> wrote:
Sorry that was an old question and here was the answer.
---------------------------------------------------------- As I had understood it (and had written in the documentation) <http://www.rbvi.ucsf.edu/chimera/docs/ContributedSoftware/morph/morph.html#p...>:
"HET residues such as ligands and ions are only included if they are present in both structures and attached to the same atom(s) in the paired chains by at least one "covalent" bond (which can be an unrealistic bond added manually, e.g., with the command bond, and subsequently undisplayed) or ion coordination pseudobond.
Once residues are paired, atoms in common within those residues are paired. In paired residues of the same type, atom pairing is straightforward. In paired residues of different types, only atoms with the same names are paired, and only a single connected fragment is kept per residue."
I hope this helps, Elaine ---------- Elaine C. Meng, Ph.D. UCSF Chimera(X) team Department of Pharmaceutical Chemistry University of California, San Francisco
-----Original Message----- From: Chimera-users [mailto:chimera-users-bounces@cgl.ucsf.edu] On Behalf Of Hernando J Sosa Sent: Sunday, September 17, 2017 8:00 PM To: chimera List Subject: [Chimera-users] morph & ligands
[This sender failed our fraud detection checks and may not be who they appear to be. Learn about spoofing at http://aka.ms/LearnAboutSpoofing]
Is there a way to keep & show the ligands in the Morph between structures?. In the morphs that I've tried the ligands do not show up.
Thanks
H.
participants (3)
-
 Elaine Meng
Elaine Meng -
 Hernando J Sosa
Hernando J Sosa -
 simon chapman
simon chapman