Surface not including all molecules?

Hello I'm trying to create a "cavity" model for visualization, I've done it before (a long time ago) and it worked great. But this time I'm having some problems. When trying with all molecules, I got some error regarding the calculation, so I split the model up into pieces and now it almost works. The problem I'm having is when I try creating a surface of one of the molecular species, I only get three "blobs" and not one big surface for all molecules. Any suggestions are most welcome. Don't know if it works, but I'll attach the PDB file for the molecules not being displayed as a surface properly.

Hi Gustaf, Your "structure" appears to be many separate small molecules. In that case, you would probably have to tell Chimera that they should be enclosed together, since usually people would want different surfaces for the different molecules. That could be done with the command surfcat. Assuming you want all the atoms to be lumped together, something like: surfcat mystuff # surface mystuff However, that surface calculation is likely to be problematic (there may be numerical failures) because the structure is very open, i.e. loosely packed: there are many holes and cavities if all those atoms are put together in one surface category. When I tried it, I did get a surface plus a warning message. One big surface was calculated successfully, but the calculation for other completely enclosed bubbles failed numerically. The big surface does show many holes and a large interior cavity that is open to the exterior. If you got that and it shows what you want, you are done. However, if you needed the additional small bubbles, or you didn't get any surface at all when you tried this, you would need to try some tweaks to avoid the numerical failure. Unfortunately a numerical failure is specific to everything (molecule, computer, all surface parameters, possibly even molecule orientation) and there is no one recipe to fix it in all situations. Some things to try are changing the atomic VDW radii slightly and/or the molecular surface vertex density or probe radius. For example, choose Favorites... Preferences, in Preferences go to Category: New Surfaces, and change settings before trying to calculate a surface. To change vdw radii, could be something like command: vdwdefine +.01 or to go back to default radii, ~vdwdefine The "split" command isn't relevant here, as it splits chains into separate models, but all the atoms in this structure are apparently chain "X". If you did have multiple chains, but you wanted all the atoms to be in one surface, you still wouldn't want to split, because that would force calculation of a separate surface for each chain. I hope this helps, Elaine ----- Elaine C. Meng, Ph.D. UCSF Computer Graphics Lab (Chimera team) and Babbitt Lab Department of Pharmaceutical Chemistry University of California, San Francisco On Dec 16, 2011, at 12:54 AM, Gustaf Olsson wrote:
Hello I'm trying to create a "cavity" model for visualization, I've done it before (a long time ago) and it worked great. But this time I'm having some problems. When trying with all molecules, I got some error regarding the calculation, so I split the model up into pieces and now it almost works.
The problem I'm having is when I try creating a surface of one of the molecular species, I only get three "blobs" and not one big surface for all molecules. Any suggestions are most welcome.
Don't know if it works, but I'll attach the PDB file for the molecules not being displayed as a surface properly.
<shell_pyr.pdb> ______________________________ Gustaf Olsson p://plato.cgl.ucsf.edu/mailman/listinfo/chimera-users
Thank you very much! I'll try the suggested command. It does not have to be a perfect surface, I just used this approach to make illustrations for presentations to illustrate a cavity in a polymer. I have 4 other types of molecules but when I tried to create the surface for all molecules I got the numerical error, that's why I singled out the pyridine residues since they where causing the error. Thank you very much for the help! // Gustaf Skickat från min iPhone 16 dec 2011 kl. 20:26 skrev "Elaine Meng" <meng@cgl.ucsf.edu>:
Hi Gustaf, Your "structure" appears to be many separate small molecules. In that case, you would probably have to tell Chimera that they should be enclosed together, since usually people would want different surfaces for the different molecules. That could be done with the command surfcat. Assuming you want all the atoms to be lumped together, something like:
surfcat mystuff # surface mystuff
However, that surface calculation is likely to be problematic (there may be numerical failures) because the structure is very open, i.e. loosely packed: there are many holes and cavities if all those atoms are put together in one surface category.
When I tried it, I did get a surface plus a warning message. One big surface was calculated successfully, but the calculation for other completely enclosed bubbles failed numerically. The big surface does show many holes and a large interior cavity that is open to the exterior. If you got that and it shows what you want, you are done.
However, if you needed the additional small bubbles, or you didn't get any surface at all when you tried this, you would need to try some tweaks to avoid the numerical failure.
Unfortunately a numerical failure is specific to everything (molecule, computer, all surface parameters, possibly even molecule orientation) and there is no one recipe to fix it in all situations. Some things to try are changing the atomic VDW radii slightly and/or the molecular surface vertex density or probe radius.
For example, choose Favorites... Preferences, in Preferences go to Category: New Surfaces, and change settings before trying to calculate a surface. To change vdw radii, could be something like command:
vdwdefine +.01
or to go back to default radii,
~vdwdefine
The "split" command isn't relevant here, as it splits chains into separate models, but all the atoms in this structure are apparently chain "X". If you did have multiple chains, but you wanted all the atoms to be in one surface, you still wouldn't want to split, because that would force calculation of a separate surface for each chain.
I hope this helps, Elaine ----- Elaine C. Meng, Ph.D. UCSF Computer Graphics Lab (Chimera team) and Babbitt Lab Department of Pharmaceutical Chemistry University of California, San Francisco
On Dec 16, 2011, at 12:54 AM, Gustaf Olsson wrote:
Hello I'm trying to create a "cavity" model for visualization, I've done it before (a long time ago) and it worked great. But this time I'm having some problems. When trying with all molecules, I got some error regarding the calculation, so I split the model up into pieces and now it almost works.
The problem I'm having is when I try creating a surface of one of the molecular species, I only get three "blobs" and not one big surface for all molecules. Any suggestions are most welcome.
Don't know if it works, but I'll attach the PDB file for the molecules not being displayed as a surface properly.
<shell_pyr.pdb> ______________________________ Gustaf Olsson p://plato.cgl.ucsf.edu/mailman/listinfo/chimera-users
It work, just a little to well. When I try I only get the "outer" surface for all molecules. Either trying to find a way to make the criteria more strict to leave more blank space "within" the sphere. Or divide the PYR residues into "clusters", creating surfaces for "aggregated" groups of molecules! But thanks a lot for the startingpoint! Best regards // Gustaf On Dec 16, 2011, at 19:37 , Elaine Meng wrote:
Hi Gustaf, Your "structure" appears to be many separate small molecules. In that case, you would probably have to tell Chimera that they should be enclosed together, since usually people would want different surfaces for the different molecules. That could be done with the command surfcat. Assuming you want all the atoms to be lumped together, something like:
surfcat mystuff # surface mystuff
However, that surface calculation is likely to be problematic (there may be numerical failures) because the structure is very open, i.e. loosely packed: there are many holes and cavities if all those atoms are put together in one surface category.
When I tried it, I did get a surface plus a warning message. One big surface was calculated successfully, but the calculation for other completely enclosed bubbles failed numerically. The big surface does show many holes and a large interior cavity that is open to the exterior. If you got that and it shows what you want, you are done.
However, if you needed the additional small bubbles, or you didn't get any surface at all when you tried this, you would need to try some tweaks to avoid the numerical failure.
Unfortunately a numerical failure is specific to everything (molecule, computer, all surface parameters, possibly even molecule orientation) and there is no one recipe to fix it in all situations. Some things to try are changing the atomic VDW radii slightly and/or the molecular surface vertex density or probe radius.
For example, choose Favorites... Preferences, in Preferences go to Category: New Surfaces, and change settings before trying to calculate a surface. To change vdw radii, could be something like command:
vdwdefine +.01
or to go back to default radii,
~vdwdefine
The "split" command isn't relevant here, as it splits chains into separate models, but all the atoms in this structure are apparently chain "X". If you did have multiple chains, but you wanted all the atoms to be in one surface, you still wouldn't want to split, because that would force calculation of a separate surface for each chain.
I hope this helps, Elaine ----- Elaine C. Meng, Ph.D. UCSF Computer Graphics Lab (Chimera team) and Babbitt Lab Department of Pharmaceutical Chemistry University of California, San Francisco
On Dec 16, 2011, at 12:54 AM, Gustaf Olsson wrote:
Hello I'm trying to create a "cavity" model for visualization, I've done it before (a long time ago) and it worked great. But this time I'm having some problems. When trying with all molecules, I got some error regarding the calculation, so I split the model up into pieces and now it almost works.
The problem I'm having is when I try creating a surface of one of the molecular species, I only get three "blobs" and not one big surface for all molecules. Any suggestions are most welcome.
Don't know if it works, but I'll attach the PDB file for the molecules not being displayed as a surface properly.
<shell_pyr.pdb> ______________________________ Gustaf Olsson p://plato.cgl.ucsf.edu/mailman/listinfo/chimera-users
Hi Gustaf, Another possibility is to simulate a density map from the atoms and show an isosurface of that density map. For example, command molmap :pyr 2.5 gridSpacing 0.3 would simulate a density map from the atoms in your file and open the Volume Viewer tool for adjusting the density contour level using the vertical bar on the histogram. The possible disadvantage is that the surface is defined differently than the molecular solvent-excluded surface, and there are adjustable parameters: the contour level, and the "resolution" (2.5 in the example commands, you could try different values) used to simulate the density map. Smaller gridSpacing gives a smoother contour surface. The molmap command has several additional options: <http://www.cgl.ucsf.edu/chimera/docs/UsersGuide/midas/molmap.html> Volume Viewer docs: <http://www.cgl.ucsf.edu/chimera/docs/ContributedSoftware/volumeviewer/framev...> Best, Elaine On Dec 16, 2011, at 12:52 PM, Gustaf Olsson wrote:
It work, just a little to well. When I try I only get the "outer" surface for all molecules. Either trying to find a way to make the criteria more strict to leave more blank space "within" the sphere. Or divide the PYR residues into "clusters", creating surfaces for "aggregated" groups of molecules!
But thanks a lot for the startingpoint!
Best regards // Gustaf
On Dec 16, 2011, at 19:37 , Elaine Meng wrote:
Hi Gustaf, Your "structure" appears to be many separate small molecules. In that case, you would probably have to tell Chimera that they should be enclosed together, since usually people would want different surfaces for the different molecules. That could be done with the command surfcat. Assuming you want all the atoms to be lumped together, something like:
surfcat mystuff # surface mystuff
However, that surface calculation is likely to be problematic (there may be numerical failures) because the structure is very open, i.e. loosely packed: there are many holes and cavities if all those atoms are put together in one surface category.
When I tried it, I did get a surface plus a warning message. One big surface was calculated successfully, but the calculation for other completely enclosed bubbles failed numerically. The big surface does show many holes and a large interior cavity that is open to the exterior. If you got that and it shows what you want, you are done.
However, if you needed the additional small bubbles, or you didn't get any surface at all when you tried this, you would need to try some tweaks to avoid the numerical failure.
Unfortunately a numerical failure is specific to everything (molecule, computer, all surface parameters, possibly even molecule orientation) and there is no one recipe to fix it in all situations. Some things to try are changing the atomic VDW radii slightly and/or the molecular surface vertex density or probe radius.
For example, choose Favorites... Preferences, in Preferences go to Category: New Surfaces, and change settings before trying to calculate a surface. To change vdw radii, could be something like command:
vdwdefine +.01
or to go back to default radii,
~vdwdefine
The "split" command isn't relevant here, as it splits chains into separate models, but all the atoms in this structure are apparently chain "X". If you did have multiple chains, but you wanted all the atoms to be in one surface, you still wouldn't want to split, because that would force calculation of a separate surface for each chain.
I hope this helps, Elaine ----- Elaine C. Meng, Ph.D. UCSF Computer Graphics Lab (Chimera team) and Babbitt Lab Department of Pharmaceutical Chemistry University of California, San Francisco
On Dec 16, 2011, at 12:54 AM, Gustaf Olsson wrote:
Hello I'm trying to create a "cavity" model for visualization, I've done it before (a long time ago) and it worked great. But this time I'm having some problems. When trying with all molecules, I got some error regarding the calculation, so I split the model up into pieces and now it almost works.
The problem I'm having is when I try creating a surface of one of the molecular species, I only get three "blobs" and not one big surface for all molecules. Any suggestions are most welcome.
Don't know if it works, but I'll attach the PDB file for the molecules not being displayed as a surface properly.
<shell_pyr.pdb> ______________________________ Gustaf Olsson p://plato.cgl.ucsf.edu/mailman/listinfo/chimera-users
_______________________________________________ Chimera-users mailing list Chimera-users@cgl.ucsf.edu http://plato.cgl.ucsf.edu/mailman/listinfo/chimera-users

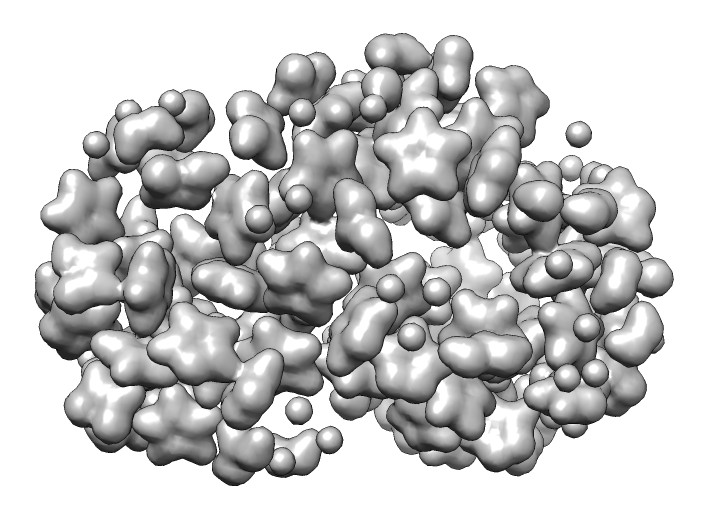
Hi Gustaf, Using the molmap command to create a density map of your molecules with resolution 2 Angstroms produces a good deal of space as shown in the attached picture. molmap #0 2 grid .2 The "grid .2" option says to make the density map grid spacing 0.2 Angstroms which gives smoother appearance than the default which is to use 1/3 the resolution. Tom Gustaf Olsson wrote:
It work, just a little to well. When I try I only get the "outer" surface for all molecules. Either trying to find a way to make the criteria more strict to leave more blank space "within" the sphere. Or divide the PYR residues into "clusters", creating surfaces for "aggregated" groups of molecules!
But thanks a lot for the startingpoint!
Best regards // Gustaf
On Dec 16, 2011, at 19:37 , Elaine Meng wrote:
Hi Gustaf, Your "structure" appears to be many separate small molecules. In that case, you would probably have to tell Chimera that they should be enclosed together, since usually people would want different surfaces for the different molecules. That could be done with the command surfcat. Assuming you want all the atoms to be lumped together, something like:
surfcat mystuff # surface mystuff
However, that surface calculation is likely to be problematic (there may be numerical failures) because the structure is very open, i.e. loosely packed: there are many holes and cavities if all those atoms are put together in one surface category.
When I tried it, I did get a surface plus a warning message. One big surface was calculated successfully, but the calculation for other completely enclosed bubbles failed numerically. The big surface does show many holes and a large interior cavity that is open to the exterior. If you got that and it shows what you want, you are done.
However, if you needed the additional small bubbles, or you didn't get any surface at all when you tried this, you would need to try some tweaks to avoid the numerical failure.
Unfortunately a numerical failure is specific to everything (molecule, computer, all surface parameters, possibly even molecule orientation) and there is no one recipe to fix it in all situations. Some things to try are changing the atomic VDW radii slightly and/or the molecular surface vertex density or probe radius.
For example, choose Favorites... Preferences, in Preferences go to Category: New Surfaces, and change settings before trying to calculate a surface. To change vdw radii, could be something like command:
vdwdefine +.01
or to go back to default radii,
~vdwdefine
The "split" command isn't relevant here, as it splits chains into separate models, but all the atoms in this structure are apparently chain "X". If you did have multiple chains, but you wanted all the atoms to be in one surface, you still wouldn't want to split, because that would force calculation of a separate surface for each chain.
I hope this helps, Elaine ----- Elaine C. Meng, Ph.D. UCSF Computer Graphics Lab (Chimera team) and Babbitt Lab Department of Pharmaceutical Chemistry University of California, San Francisco
On Dec 16, 2011, at 12:54 AM, Gustaf Olsson wrote:
Hello I'm trying to create a "cavity" model for visualization, I've done it before (a long time ago) and it worked great. But this time I'm having some problems. When trying with all molecules, I got some error regarding the calculation, so I split the model up into pieces and now it almost works.
The problem I'm having is when I try creating a surface of one of the molecular species, I only get three "blobs" and not one big surface for all molecules. Any suggestions are most welcome.
Don't know if it works, but I'll attach the PDB file for the molecules not being displayed as a surface properly.
<shell_pyr.pdb> ______________________________ Gustaf Olsson p://plato.cgl.ucsf.edu/mailman/listinfo/chimera-users
_______________________________________________ Chimera-users mailing list Chimera-users@cgl.ucsf.edu http://plato.cgl.ucsf.edu/mailman/listinfo/chimera-users
{kind=link}

Hello, it seems that if some amber trajectory but also PDB containing solvated structure/s is loaded into Chimera, there is impossible to select by residue number (using command SELECT in command line or by dialog SELECT->ATOM SPECIFIER) any solvent reside (at least in case of WAT solvent). Am I right ? Perhaps it is possible but maybe with some more complicated command than the usual select :N where N is the residue number. Thanks in advance for any comments ! Best wishes, Marek PS: I am using alpha version from 18th November, but I remember this problem also from previous versions.
Hello, I just found solution (at least in case of water solvent :)) ). When one want to select water residue with number N it is necessary do it like this: select :N.water (not just "select :N" which works in case of solute or ions ) Best wishes, Marek Dne Tue, 20 Dec 2011 03:27:12 +0100 Marek Maly <marek.maly@ujep.cz> napsal/-a:
Hello, it seems that if some amber trajectory but also PDB containing solvated structure/s is loaded into Chimera, there is impossible to select by residue number (using command SELECT in command line or by dialog SELECT->ATOM SPECIFIER) any solvent reside (at least in case of WAT solvent).
Am I right ?
Perhaps it is possible but maybe with some more complicated command than the usual
select :N
where N is the residue number.
Thanks in advance for any comments !
Best wishes,
Marek
PS: I am using alpha version from 18th November, but I remember this problem also from previous versions.
_______________________________________________ Chimera-users mailing list Chimera-users@cgl.ucsf.edu http://plato.cgl.ucsf.edu/mailman/listinfo/chimera-users
__________ Informace od ESET NOD32 Antivirus, verze databaze 6725 (20111219) __________
Tuto zpravu proveril ESET NOD32 Antivirus.
-- Tato zpráva byla vytvořena převratným poštovním klientem Opery: http://www.opera.com/mail/
Hi Marek, Without an example, I could only suggest trying something like select :515.water or select :515 & solvent If that doesn't work, I would need an example file (could send to just me if you don't want to send to the whole list). Best, Elaine ---------- Elaine C. Meng, Ph.D. UCSF Computer Graphics Lab (Chimera team) and Babbitt Lab Department of Pharmaceutical Chemistry University of California, San Francisco On Dec 19, 2011, at 6:27 PM, Marek Maly wrote:
Hello, it seems that if some amber trajectory but also PDB containing solvated structure/s is loaded into Chimera, there is impossible to select by residue number (using command SELECT in command line or by dialog SELECT->ATOM SPECIFIER) any solvent reside (at least in case of WAT solvent).
Am I right ?
Perhaps it is possible but maybe with some more complicated command than the usual
select :N
where N is the residue number.
Thanks in advance for any comments !
Best wishes,
Marek
PS: I am using alpha version from 18th November, but I remember this problem also from previous versions.
participants (4)
-
 Elaine Meng
Elaine Meng -
 Gustaf Olsson
Gustaf Olsson -
 Marek Maly
Marek Maly -
 Tom Goddard
Tom Goddard