
Hi Alexander, I have talked to someone (forgot name, at Grenoble EM fitting workshop a year ago) who was interested in computing SAXS profiles for EM blobs of unknown proteins and your EM2DAM program could make that possible. It might also have utility in analyzing EM maps. I wonder about details like how how far apart are the dummy atoms placed. Is documentation for the program available? About 9 months ago I added to Chimera the ability to compute a SAXS profile from an atomic model which is then plotted against an experimental profile. The idea is that you could test various atomic model conformations to see which is most compatible with SAXS data. Since then YZ in our lab has added new options and made it work using a web service in addition to the option of running a local program. This is in Chimera daily builds but not in the 1 year old Chimera 1.4 production release. We will have a Chimera 1.5 release soon that will have it. It uses FoXS (http://modbase.compbio.ucsf.edu/foxs/about.html) for the profile computation. Here's a description from the Chimera User's Guide of the Chimera user interface. http://www.cgl.ucsf.edu/chimera/docs/ContributedSoftware/saxs/saxs.html Tom
Hi Tom
First of all, thank you very much for very helpful comments. Although thanks for such a nice tool as Chimera!
The reason I was asking all questions about volume viewing and MRC file format is that I am developing a tool that converts EM density maps to dummy atom models (EM2DAM). Bead models (PDB format) can be used by other software to validate EM models for example.
You can read more here: http://www.mail-archive.com/ccp4bb@jiscmail.ac.uk/msg17870.html
I like Chimera a lot. And if you be interested in integrating/using em2dam (fortran90) in Chimera, let me know.
Btw, could you comment on SAXS function in Chimera?
Kind regards from Hamburg Alexander

Just a quick mention that the documentation Tom mentioned below for the SAXS tool in Chimera is a little out of sync with the daily build because the dialog is in the process of being modified. In general, however, the descriptions in that page and the FoXS page (linked to that page) will explain the options. Elaine On Oct 15, 2010, at 4:26 PM, Tom Goddard wrote:
Here's a description from the Chimera User's Guide of the Chimera user interface.
http://www.cgl.ucsf.edu/chimera/docs/ContributedSoftware/saxs/saxs.html

Hi Tom Dummy atoms are placed according to pixel size. The only documentation is available from "em2dam -help". You can get example mentioned in help from EMDB. I am sending you linux binary. We are not using macs here much. I hope this is not a problem. In attachment is a test version. There are some bugs I work on at the moment, but feedback is very welcome. It is very interesting, because I developed last version of CRYSOL (2.7). I never used FOXS though. What you plan to do is actually validating high resolution models (NMR or Xray). Actually how would you treat NMR models? Often I see that not low energy (first conformer), but other models fit SAXS data better, or even their combination. Plausible addition to it would be validation of EM models by first generating dummy models and then calculating theoretical curve and fitting to experimental data.
http://www.cgl.ucsf.edu/chimera/docs/ContributedSoftware/saxs/saxs.htm l
I am actually using alpha version 1.5 (build 31455). I tried a simple test, namely, loaded lysozyme and fit it to experimental data. The fit is very bad! and why q is plotted starting from 0.2? Kind regards Alex On Saturday 16 October 2010 01:26:50 you wrote:
Hi Alexander,
I have talked to someone (forgot name, at Grenoble EM fitting workshop a year ago) who was interested in computing SAXS profiles for EM blobs of unknown proteins and your EM2DAM program could make that possible. It might also have utility in analyzing EM maps. I wonder about details like how how far apart are the dummy atoms placed. Is documentation for the program available?
About 9 months ago I added to Chimera the ability to compute a SAXS profile from an atomic model which is then plotted against an experimental profile. The idea is that you could test various atomic model conformations to see which is most compatible with SAXS data. Since then YZ in our lab has added new options and made it work using a web service in addition to the option of running a local program. This is in Chimera daily builds but not in the 1 year old Chimera 1.4 production release. We will have a Chimera 1.5 release soon that will have it. It uses FoXS (http://modbase.compbio.ucsf.edu/foxs/about.html) for the profile computation. Here's a description from the Chimera User's Guide of the Chimera user interface.
http://www.cgl.ucsf.edu/chimera/docs/ContributedSoftware/saxs/saxs.htm l
Tom
-- Alexander Shkumatov, predoc -- Biological Small Angle Scattering Group EMBL Hamburg Outstation c/o DESY -- Building 25A Notkestraße 85 -- 22603 Hamburg -- Germany tel: +49 40 89902 178 fax: +49 40 89902 149 Email: ashkumat@embl-hamburg.de
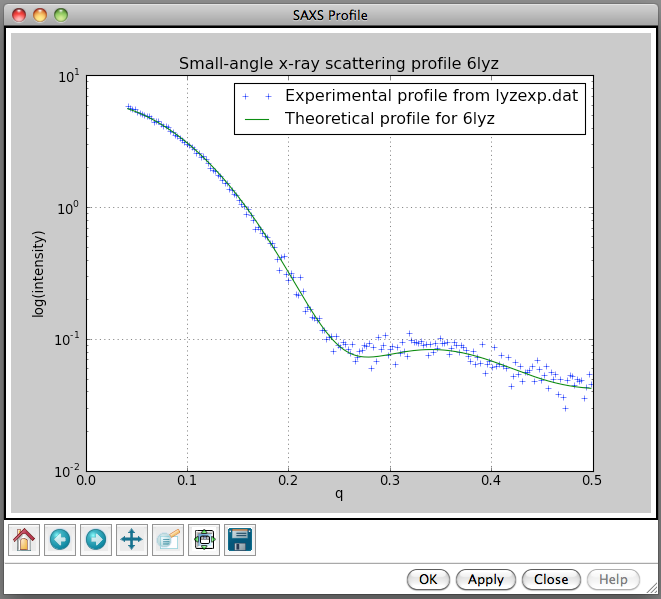
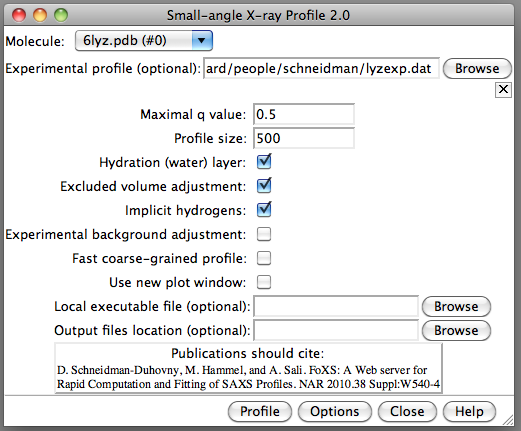
Hi Alex, I'll give em2dam a try. I agree it would be useful for comparing EM and SAXS data. For NMR models I think it would be useful to generate SAXS profiles for each conformer and see which fit the experimental data the best. Probably could plot profiles for 20 conformers on one plot to get an idea of the degree of variation. Sometimes the NMR conformers indicate flexibility in solution and maybe some combination of them could provide a good fit to the SAXS data in that case. But other times, the variation among conformers is just due to lack of NMR constraints and is not due to flexibility in solution. There is a strong risk of over-fitting if multiple NMR conformers are used to fit SAXS data. Not sure why your test of the Chimera SAXS profile calculation did not work well for lyzosome. I've attached two images using Chimera daily build 31662 (Oct 17) where it worked fine for lyzosome with the experimental profile I have. Maybe the minimum q = 0.2 is related to your experimental profile not having small q values? YZ in our lab works on the Chimera SAXS tool and could give a better answer. Tom
Hi Tom
Dummy atoms are placed according to pixel size. The only documentation is available from "em2dam -help". You can get example mentioned in help from EMDB.
I am sending you linux binary. We are not using macs here much. I hope this is not a problem. In attachment is a test version. There are some bugs I work on at the moment, but feedback is very welcome.
It is very interesting, because I developed last version of CRYSOL (2.7). I never used FOXS though. What you plan to do is actually validating high resolution models (NMR or Xray). Actually how would you treat NMR models? Often I see that not low energy (first conformer), but other models fit SAXS data better, or even their combination. Plausible addition to it would be validation of EM models by first generating dummy models and then calculating theoretical curve and fitting to experimental data.
http://www.cgl.ucsf.edu/chimera/docs/ContributedSoftware/saxs/saxs.htm l
I am actually using alpha version 1.5 (build 31455). I tried a simple test, namely, loaded lysozyme and fit it to experimental data. The fit is very bad! and why q is plotted starting from 0.2?
Kind regards Alex
On Saturday 16 October 2010 01:26:50 you wrote:
Hi Alexander,
I have talked to someone (forgot name, at Grenoble EM fitting workshop a year ago) who was interested in computing SAXS profiles for EM blobs of unknown proteins and your EM2DAM program could make that possible. It might also have utility in analyzing EM maps. I wonder about details like how how far apart are the dummy atoms placed. Is documentation for the program available?
About 9 months ago I added to Chimera the ability to compute a SAXS profile from an atomic model which is then plotted against an experimental profile. The idea is that you could test various atomic model conformations to see which is most compatible with SAXS data. Since then YZ in our lab has added new options and made it work using a web service in addition to the option of running a local program. This is in Chimera daily builds but not in the 1 year old Chimera 1.4 production release. We will have a Chimera 1.5 release soon that will have it. It uses FoXS (http://modbase.compbio.ucsf.edu/foxs/about.html) for the profile computation. Here's a description from the Chimera User's Guide of the Chimera user interface.
http://www.cgl.ucsf.edu/chimera/docs/ContributedSoftware/saxs/saxs.htm l
Tom
_______________________________________________ Chimera-users mailing list Chimera-users@cgl.ucsf.edu http://plato.cgl.ucsf.edu/mailman/listinfo/chimera-users
{kind=link}
{kind=link}

Hi Tom I think I found the problem: units in my data are reverse nm, whereas by default angstroms are taken. It would be nice to have "choose units" in options. Alex
Not sure why your test of the Chimera SAXS profile calculation did not work well for lyzosome. I've attached two images using Chimera daily build 31662 (Oct 17) where it worked fine for lyzosome with the experimental profile I have. Maybe the minimum q = 0.2 is related to your experimental profile not having small q values? YZ in our lab works on the Chimera SAXS tool and could give a better answer.
Tom
Alexander Shkumatov, predoc -- Biological Small Angle Scattering Group EMBL Hamburg Outstation c/o DESY -- Building 25A Notkestraße 85 -- 22603 Hamburg -- Germany tel: +49 40 89902 178 fax: +49 40 89902 149 Email: shkumatau@embl-hamburg.de ----------------------------------------------------------------------
Hi Alex, Yes, we need an option to choose 1/nm or 1/Angstrom for q units of the experimental SAXS profile. I'll ask YZ who works on the Chimera SAXS dialog if he can add that. Tom
Hi Tom I think I found the problem: units in my data are reverse nm, whereas by default angstroms are taken. It would be nice to have "choose units" in options. Alex
Not sure why your test of the Chimera SAXS profile calculation did not work well for lyzosome. I've attached two images using Chimera daily build 31662 (Oct 17) where it worked fine for lyzosome with the experimental profile I have. Maybe the minimum q = 0.2 is related to your experimental profile not having small q values? YZ in our lab works on the Chimera SAXS tool and could give a better answer.
Tom
Alexander Shkumatov, predoc -- Biological Small Angle Scattering Group EMBL Hamburg Outstation c/o DESY -- Building 25A Notkestraße 85 -- 22603 Hamburg -- Germany
----------------------------------------------------------------------

Alex, We could add the nm/Angstrom option. Would you please send me your structure file and experimental data so we could test our codes. Thanks, YZ On Oct 19, 2010, at 11:06 AM, Tom Goddard wrote:
Hi Alex,
Yes, we need an option to choose 1/nm or 1/Angstrom for q units of the experimental SAXS profile. I'll ask YZ who works on the Chimera SAXS dialog if he can add that.
Tom
Hi Tom I think I found the problem: units in my data are reverse nm, whereas by default angstroms are taken. It would be nice to have "choose units" in options. Alex
Not sure why your test of the Chimera SAXS profile calculation did not work well for lyzosome. I've attached two images using Chimera daily build 31662 (Oct 17) where it worked fine for lyzosome with the experimental profile I have. Maybe the minimum q = 0.2 is related to your experimental profile not having small q values? YZ in our lab works on the Chimera SAXS tool and could give a better answer.
Tom
Alexander Shkumatov, predoc -- Biological Small Angle Scattering Group EMBL Hamburg Outstation c/o DESY -- Building 25A Notkestraße 85 -- 22603 Hamburg -- Germany
----------------------------------------------------------------------
_______________________________________________ Chimera-users mailing list Chimera-users@cgl.ucsf.edu http://plato.cgl.ucsf.edu/mailman/listinfo/chimera-users
participants (5)
-
 Alex Shkumatov
Alex Shkumatov -
 Alex Shkumatov
Alex Shkumatov -
 Elaine Meng
Elaine Meng -
 Tom Goddard
Tom Goddard -
 Yang, Zheng(YZ)
Yang, Zheng(YZ)