Electrostatic Potential based on Poisson-Boltzmann using APBS

Dear chimera users, I am using chimera 1.10.1. I did following steps to obtain Electrostatic Potential based on Poisson-Boltzmann using APBS: open... pr.pdb action... surface... show to obtain *.dx file, I typed APBS in command line, after minutes, *.dx file was created and surface color window was opened, I pressed color tab. Please see the figure in following link: https://www.dropbox.com/s/99gjecypqu910hu/apbs.png?dl=0 Why are the colors black and white in the surface, instead of blue and red? Any help will highly appreciated.
Also, I used surfvalues.py script. After opening the this script, I encountered with: tmpgi8ixd.dx value at surface point = nan When I put mouse cursor on the different points of surface, no potential value are seen. Why is the reason of this state? Best, Leila

Hi Leila, Well, “nan” generally means the value is out of range, so one possibility is that your map from APBS does not cover that part of the surface. If you did coloring, it might also show this. Besides a problem with the map or its placement relative to the surface, a second possiblity is that the script is too old and has stopped working. However, the feature was added to Chimera in 2011, so no reason to use that script anyway. Just show the Options in the Surface Color (Electrostatic Surface Coloring) tool and turn on "Report value at mouse position”: <http://www.rbvi.ucsf.edu/chimera/docs/ContributedSoftware/surfcolor/surfcolo...> I just tried this option. After I clicked Color to apply the coloring, the value-reporting works fine for me, giving the potential value and the XYZ coordinates of the surface point in the status line at the bottom of the Chimera window. I hope this helps, Elaine ----- Elaine C. Meng, Ph.D. UCSF Computer Graphics Lab (Chimera team) and Babbitt Lab Department of Pharmaceutical Chemistry University of California, San Francisco On Jul 17, 2015, at 1:40 AM, leila karami <karami.leila1@gmail.com> wrote:
Also, I used surfvalues.py script. After opening the this script, I encountered with:
tmpgi8ixd.dx value at surface point = nan
When I put mouse cursor on the different points of surface, no potential value are seen.
Why is the reason of this state?
Best, Leila
Dear Elaine, Thanks for your answer. 1) I turned on "Report value at mouse position" in Surface Color (Electrostatic Surface Coloring) tool. After, I clicked Color to apply the coloring, the value-reporting works fine for me, giving the potential value and the XYZ coordinates of the surface point in the status line at the bottom of the Chimera window. I used this scripts from the following tutorial: http://www.cgl.ucsf.edu/chimera/videodoc/SurfaceValues/index.html Please note to the time 3:20 in this video in which there is following line in the status line at the bottom of the Chimera window. *.dx value at surface point = 0.78023. What is the reason of this state? How to have value at surface point instead of XYZ coordinates of the surface point. 2) Please answer my first question in mailing list: I did following steps to obtain Electrostatic Potential based on Poisson-Boltzmann using APBS: open... pr.pdb action... surface... show to obtain *.dx file, I typed APBS in command line, after minutes, *.dx file was created and surface color window was opened, I pressed color tab. Please see the figure in following link: https://www.dropbox.com/s/99gjecypqu910hu/apbs.png?dl=0 Why are the colors black and white in the surface, instead of blue and red?
Hi Leila, (1) The “report values” option already gives you the potential value. It gives 4 values: electrostatic potential, x coord, y coord, z coord. In general, videos may show an older version of a script and/or Chimera, so things may be different now. We try to keep the “User’s Guide” HTML documentation that is connected to the Help menu current, but other types of documentation may be for older versions. (2) Usually a picture isn’t enough information, we would need the data to tell for sure (the dx file and the PDB file). However, I’m pretty sure it is a problem with your map values, which are the first things to check when the coloring is weird. Does mouseover report reasonable potential values (mostly small negative and small positive)? If your values are not reasonable, for example “nan”, then it is a problem with the map. Probably you did not add hydrogens and charges before running APBS. It is necessary to do both of those things first, and also other structure cleanup may be required. There are few options for which tools you use for these steps, so I refer you to the manual for more information and links: <http://www.rbvi.ucsf.edu/chimera/docs/UsersGuide/midas/apbs.html> The APBS graphical interface in Chimera does check for charges and complains if you try to run it without them. However, the “apbs” command does not do this error checking. Most people use the graphical interface first, then switch to the command for scripting after they know the process. One option for processing before APBS is PDB2PQR (in menu under Tools… Structure Editing, or command “pdb2pqr”). PDB2PQR will open an additional copy of the structure that has charges. You would then need to run APBS (in menu under Tools.. Surface /Binding Analysis, or command “apbs”) on the NEW copy of the molecule, not the original copy that still doesn’t have charges. I hope this helps, Elaine ----- Elaine C. Meng, Ph.D. UCSF Computer Graphics Lab (Chimera team) and Babbitt Lab Department of Pharmaceutical Chemistry University of California, San Francisco On Jul 17, 2015, at 8:35 AM, leila karami <karami.leila1@gmail.com> wrote:
Dear Elaine,
Thanks for your answer.
1) I turned on "Report value at mouse position" in Surface Color (Electrostatic Surface Coloring) tool.
After, I clicked Color to apply the coloring, the value-reporting works fine for me, giving the potential value and the XYZ coordinates of the surface point in the status line at the bottom of the Chimera window.
I used this scripts from the following tutorial:
http://www.cgl.ucsf.edu/chimera/videodoc/SurfaceValues/index.html
Please note to the time 3:20 in this video in which there is following line in the status line at the bottom of the Chimera window.
*.dx value at surface point = 0.78023.
What is the reason of this state? How to have value at surface point instead of XYZ coordinates of the surface point.
2) Please answer my first question in mailing list:
I did following steps to obtain Electrostatic Potential based on Poisson-Boltzmann using APBS:
open... pr.pdb action... surface... show to obtain *.dx file, I typed APBS in command line, after minutes, *.dx file was created and surface color window was opened, I pressed color tab.
Please see the figure in following link: https://www.dropbox.com/s/99gjecypqu910hu/apbs.png?dl=0
Why are the colors black and white in the surface, instead of blue and red?
Dear Elaine, 1) "report values" gives only 3 values (x coord, y coord, z coord) and not 4 values. 2) Based on your suggestion, I did following steps: Tools... structure Editing... pdb2pqr But I encountered with: Service 'opal:pdb2pqr_1.8' is unavailable. 3) Again, I tried the apbs in command line. To have my dx and the PDB files, please check the following links: https://www.dropbox.com/s/n4zv0lq7s4p1pz2/44.pdb?dl=0 https://www.dropbox.com/s/w6dg4adb498178r/tmpiwv0lf.dx?dl=0 Again, electrostatic surface is as black and white instead of red and blue. Thanks in advance, Leila
Dear Leila, As I said before, you must add charges to the structure before running APBS, or else your map will not have values and the coloring will be black and white. According to the error message, the pdb2pqr calculation did not run, so you did not have charges and your problem is exactly the same as before. The pdb2pqr service changed its URL, so we had to change Chimera, and so to run that service you need to use a newer version of Chimera, the daily build: <http://www.rbvi.ucsf.edu/chimera/download.html#daily> OR, instead of using PDB2PQR you can use the other tools mentioned in the APBS manual page (link sent in previous message) for preparing the structure, for example Dock Prep (in menu under Tools… Structure Editing), which will in turn call AddH and AddCharge tools for adding hydrogens and charges. However, the PDB2PQR charges may be better for Poisson-Boltzmann calculations, again as mentioned in the APBS manual page. Elaine On Jul 17, 2015, at 1:11 PM, leila karami <karami.leila1@gmail.com> wrote:
Dear Elaine,
1) "report values" gives only 3 values (x coord, y coord, z coord) and not 4 values.
2) Based on your suggestion, I did following steps:
Tools... structure Editing... pdb2pqr
But I encountered with:
Service 'opal:pdb2pqr_1.8' is unavailable.
3) Again, I tried the apbs in command line.
To have my dx and the PDB files, please check the following links:
https://www.dropbox.com/s/n4zv0lq7s4p1pz2/44.pdb?dl=0 https://www.dropbox.com/s/w6dg4adb498178r/tmpiwv0lf.dx?dl=0
Again, electrostatic surface is as black and white instead of red and blue.
Thanks in advance, Leila
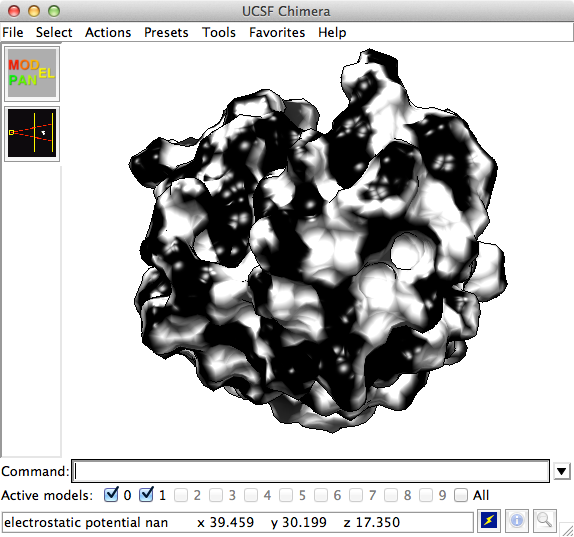
Hi Leila, Here’s an image of your structure and dx map in Chimera, with status line showing the 4 values. The ESP value is given as “nan”, meaning there is nothing useful in the map file because it was calculated without charges. Elaine On Jul 17, 2015, at 1:11 PM, leila karami <karami.leila1@gmail.com> wrote:
Dear Elaine,
1) "report values" gives only 3 values (x coord, y coord, z coord) and not 4 values.
2) Based on your suggestion, I did following steps:
Tools... structure Editing... pdb2pqr
But I encountered with:
Service 'opal:pdb2pqr_1.8' is unavailable.
3) Again, I tried the apbs in command line.
To have my dx and the PDB files, please check the following links:
https://www.dropbox.com/s/n4zv0lq7s4p1pz2/44.pdb?dl=0 https://www.dropbox.com/s/w6dg4adb498178r/tmpiwv0lf.dx?dl=0
Again, electrostatic surface is as black and white instead of red and blue.
Thanks in advance, Leila _______________________________________________ Chimera-users mailing list Chimera-users@cgl.ucsf.edu http://plato.cgl.ucsf.edu/mailman/listinfo/chimera-users
{kind=link}
Dear Elaine, Very thanks for your time and consideration. I downloaded the daily build version of chimera. After using PDB2PQR in command line, status line shows that output.pqr file was generated. Then, I used APBS in command line, dx file was created. But after coloring, the colors are as black and white. I think chimera uses a pqr file other than output.pqr file which PDB2PQR has generated. After using APBS in command line, pqr, in and dx files were created in following address: C:\Users\karami\AppData\Local\Temp\tmphrfmlq How to find output.pqr file to use that as inout for APBS? Is my manner wrong? Thanks in advance, Leila
Dear Leila, Probably the PDB file you opened is model 0 in Chimera. The PQR file is automatically opened by PDB2PQR when it finishes running, and that is probably model 1 in Chimera. You can just close the original PDB file before you run APBS, or you can make sure to tell APBS to use the new one. Either way, you can see which file is which model number by looking in the Model Panel (menu: Favorites… Model Panel). You can close a model using the Model Panel or with a command, for example: close 0 If you don’t close the original PDB file, you can still tell APBS to use the new model. In the APBS graphical interface (menu Tools… Surface/Binding Analysis… APBS), you can change the “Molecule:” setting at the top to be the new model output by PDB2PQR, or the apbs command can be something like: apbs molecule #1 (using the correct number for the PDB2PQR file, of course) This is all mentioned in the help pages, so it would be useful to read the help for what you are doing before asking this email list. For example, you can see the help for the apbs command by using the command: help apbs <http://www.rbvi.ucsf.edu/chimera/docs/ContributedSoftware/apbs/apbs.html> <http://www.rbvi.ucsf.edu/chimera/docs/UsersGuide/midas/apbs.html> Also for basic familiarity with Chimera, you could try tutorials, see menu Help… Tutorials, and also the longer “getting started” tutorial at our website: <http://www.rbvi.ucsf.edu/Outreach/Tutorials/GettingStarted.html> I hope this helps, Elaine ----- Elaine C. Meng, Ph.D. UCSF Computer Graphics Lab (Chimera team) and Babbitt Lab Department of Pharmaceutical Chemistry University of California, San Francisco On Jul 18, 2015, at 12:37 AM, leila karami <karami.leila1@gmail.com> wrote:
Dear Elaine, Very thanks for your time and consideration. I downloaded the daily build version of chimera.
After using PDB2PQR in command line, status line shows that output.pqr file was generated.
Then, I used APBS in command line, dx file was created. But after coloring, the colors are as black and white.
I think chimera uses a pqr file other than output.pqr file which PDB2PQR has generated.
After using APBS in command line, pqr, in and dx files were created in following address:
C:\Users\karami\AppData\Local\Temp\tmphrfmlq
How to find output.pqr file to use that as inout for APBS?
Is my manner wrong? Thanks in advance, Leila _
Dear Elaine, I checked the useful link. I opened pr.pdb file (#0). I used pdb2pqr. Then, a new pdb file was created. I opened this new file (#1). Then, I used apbs molecule #1. Then, I opened created dx file. Unfortunately, surface is black and white. I confused. Which part of my steps is wrong? Please guide me to resolve this problem.
Dear Leila, Your description does not make sense to me because you do not need to open the pqr file or the dx file yourself. They will both open automatically when the calculations are done. Otherwise it sounds OK, and it works fine when I do it. Maybe (especially if you opened some files yourself) there are extra models and you are confused about which number is which. You should always look in the Model Panel to make sure. Otherwise I have no more ideas. Elaine On Jul 18, 2015, at 11:23 AM, leila karami <karami.leila1@gmail.com> wrote:
Dear Elaine,
I checked the useful link.
I opened pr.pdb file (#0).
I used pdb2pqr. Then, a new pdb file was created.
I opened this new file (#1). Then, I used apbs molecule #1.
Then, I opened created dx file.
Unfortunately, surface is black and white.
I confused. Which part of my steps is wrong?
Please guide me to resolve this problem.
Dear Elaine, I opened my original pdb file as #1 model. There is a important point: you said PQR file is automatically opened by PDB2PQR when it finishes running. But in my case, generated PQR file is not opened automatically. Why? You are right. After finishing apbs, dx file is opened automatically .
Dear Elaine, Let me know those steps you exaclty did (step by step). Best wishes, Leila
On Jul 18, 2015, at 12:08 PM, leila karami <karami.leila1@gmail.com> wrote:
Dear Elaine, Let me know those steps you exaclty did (step by step). Best wishes, Leila
start Chimera you could use graphical interfaces or commands, but below are example commands open 1zik (… or instead of 1zik, your protein pdb file) pdb2pqr (… wait for job to finish, output will be opened automatically as model 1) close 0 surf apbs (…wait for job to finish, dx file will be opened automatically; use Surface Color dialog)
Dear Elaine, Very very thanks for your time and consideration. I appreciate your patience in answering to my many questions. Finally, my problem was solved. My mistake was that I opened pdb file instead of pqr file. Now pqr file was created as #1. After closing original pdb file (#0), all things is wright. Thanks in advance, Leila
If pdb2pqr runs successfully, it will open the file automatically. It does not create a pdb file, it creates a pqr file. Maybe you are looking at the Reply Log and seeing that there is a temp pdb file created. This is NOT the output. It is a temporary pdb file that is used as the input, and pdb files do not have charges in them. So I can only guess either you didn’t wait long enough, or the pdb2pqr web service failed on your structure. We have not had this problem. Elaine On Jul 18, 2015, at 11:43 AM, leila karami <karami.leila1@gmail.com> wrote:
Dear Elaine,
I opened my original pdb file as #1 model.
There is a important point:
you said PQR file is automatically opened by PDB2PQR when it finishes running. But in my case, generated PQR file is not opened automatically. Why?
You are right. After finishing apbs, dx file is opened automatically .
participants (2)
-
 Elaine Meng
Elaine Meng -
 leila karami
leila karami