
Hi Zhengzheng The format of your PDB file is not standard because all the H atoms are all at the end instead of with their their residues. Also they are all in a different chain (chain A) than the other atoms (chain B). Whatever program wrote this file wrote it incorrectly. You can easily see all atoms, e.g. commands: ~ribbon display (or use Actions menu to do the same things) but if you use Chimera features requiring chemical knowledge (findhbond etc.) it will not work correctly if the H and the other atoms are not recognized as being in the same residue. I tried editing so that all atoms were chain B, but it did not fix the problem, which is apparently instead caused by placing all the H atoms at the end instead of with their individual residues. You could fix it be removing and re-adding hydrogens in Chimera, but then the H atom positions wouldn’t be exactly the same. Commands: delete H addh It is recommended to send questions to chimera-users@cgl.ucsf.edu (CC’ed here) in case I’m not available to answer. Best, Elaine ----- Elaine C. Meng, Ph.D. UCSF Chimera(X) team Department of Pharmaceutical Chemistry University of California, San Francisco
On Nov 7, 2018, at 8:02 AM, zheng l <zhengsfriend@hotmail.com> wrote:
Dear Dr. Meng
I used Chimera software for analyzing many protein crystal structures and then modified them and it has so faithfully doing job for me. However, this structure that I attached below is very difficult to modify.
The problem is that the hydrogens in structure are hidden. Usually when I need to, for example, replaces a carbon to a nitrogen in a Phe the nitrogen will attached with a hydrogen if I set to have 3 bond for the nitrogen. But this structure is not showing hydrogens at all
Would you please help me find a way of show all hydrogens need for modifying the protein structure?
Thank you!
Best regards!
Zhengzheng
<1tup chain b opt 2.pdb>

Dear Dr. Meng. Thank you so much for suggestion of add and delete H in the command line! This time it works for the structure modification! I want to ask you one more question about an error that pop up every time i am trying to modify and I attached the error message above. (this is not a major issue but i just do not understand why it always show up whenever i set modification to the structure.) I know the error is related to the Zn atom in the structure, but p53 sequence specific DNA binding domain does contain a Zn atom that binds 4 different amino acids. I only know very little python and does not know where the error comes from. May you please suggest why is Chimera not happy with the Zn atom in the structure? Thank you! Zhengzheng ________________________________ From: Elaine Meng <meng@cgl.ucsf.edu> Sent: Wednesday, November 7, 2018 9:29 AM To: zheng l Cc: chimera-users Subject: Re: 1up opt Hi Zhengzheng The format of your PDB file is not standard because all the H atoms are all at the end instead of with their their residues. Also they are all in a different chain (chain A) than the other atoms (chain B). Whatever program wrote this file wrote it incorrectly. You can easily see all atoms, e.g. commands: ~ribbon display (or use Actions menu to do the same things) but if you use Chimera features requiring chemical knowledge (findhbond etc.) it will not work correctly if the H and the other atoms are not recognized as being in the same residue. I tried editing so that all atoms were chain B, but it did not fix the problem, which is apparently instead caused by placing all the H atoms at the end instead of with their individual residues. You could fix it be removing and re-adding hydrogens in Chimera, but then the H atom positions wouldn’t be exactly the same. Commands: delete H addh It is recommended to send questions to chimera-users@cgl.ucsf.edu (CC’ed here) in case I’m not available to answer. Best, Elaine ----- Elaine C. Meng, Ph.D. UCSF Chimera(X) team Department of Pharmaceutical Chemistry University of California, San Francisco
On Nov 7, 2018, at 8:02 AM, zheng l <zhengsfriend@hotmail.com> wrote:
Dear Dr. Meng
I used Chimera software for analyzing many protein crystal structures and then modified them and it has so faithfully doing job for me. However, this structure that I attached below is very difficult to modify.
The problem is that the hydrogens in structure are hidden. Usually when I need to, for example, replaces a carbon to a nitrogen in a Phe the nitrogen will attached with a hydrogen if I set to have 3 bond for the nitrogen. But this structure is not showing hydrogens at all
Would you please help me find a way of show all hydrogens need for modifying the protein structure?
Thank you!
Best regards!
Zhengzheng
<1tup chain b opt 2.pdb>
{kind=link}
Hi Zhengzheng, I don’t know python either, but maybe you just need to get a newer Chimera. I tried the production release (1.13) and a recent daily build (1.14) and I did not see any error message when opening your PDB file. I do see some warnings in Reply Log (open from Favorites menu) like this: warning: cannot find LINK/SSBOND residue ZN (953 ) … but it is probably just because the header information in the first part of the file refers to the other chains that I assume you had deleted from the structure. In that case the warning messages are expected and could be ignored. Best, Elaine ----- Elaine C. Meng, Ph.D. UCSF Chimera(X) team Department of Pharmaceutical Chemistry University of California, San Francisco
On Nov 7, 2018, at 7:58 PM, zheng l <zhengsfriend@hotmail.com> wrote:
Dear Dr. Meng.
Thank you so much for suggestion of add and delete H in the command line! This time it works for the structure modification!
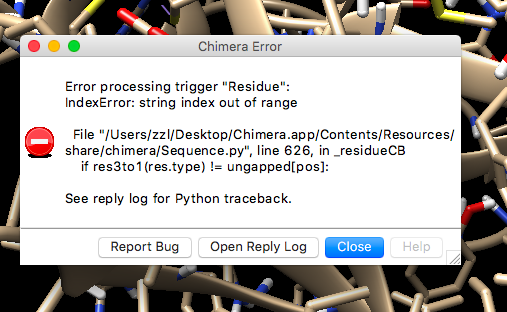
I want to ask you one more question about an error that pop up every time i am trying to modify and I attached the error message above. (this is not a major issue but i just do not understand why it always show up whenever i set modification to the structure.)
I know the error is related to the Zn atom in the structure, but p53 sequence specific DNA binding domain does contain a Zn atom that binds 4 different amino acids.
I only know very little python and does not know where the error comes from.
May you please suggest why is Chimera not happy with the Zn atom in the structure?
Thank you! Zhengzheng

Hello, Recently, I stumbled upon this open source tool: PLIP - Protein-Ligand Interaction Profiler Analyze and visualize non-covalent protein-ligand interactions in PDB files From the abstract: --- [...] detection and visualization of [...] seven interaction types (hydrogen bonds, hydrophobic contacts, pi-stacking, pi-cation interactions, salt bridges, water bridges and halogen bonds). --- Website: http://plip.biotec.tu-dresden.de Open-source code: https://github.com/ssalentin/plip Paper: https://academic.oup.com/nar/article-pdf/43/W1/W443/17435622/gkv315.pdf or https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4489249/ And, when I am inside chimera looking at a protein-ligand complex, it would be super nice if this tool could be launched from within chimera (as in a menu "Tools -> Surface/Binding Analysis -> PLIP analysis (visualize non-covalent interactions)"). Or, maybe it could be a toggable button inside of ViewDock. Best regards, Francois.

Hi! We have developed a Chimera extension for this (PLIPGUI), within our Tangram Project. It should be usable separate of the full suite, but the dependencies must be installed manually: - PLIPGUI: The extension -> https://github.com/insilichem/tangram_plipgui - libtangram: Some common code for Tangram extensions -> https://github.com/insilichem/libtangram - PLIP: The code itself, with no dependencies - Openbabel: Plip needs it. - In some cases, I had to install lxml and future, as well. I normally create a conda environment with Python 2.7 and install the dependencies there, but you can also use a normal directory and use pip install -t <location> <packages> to install them there. Then, make sure to point to that location in the Chimera> Favorites> Add to Favorites/Tools> Locations dialog. More information here: https://tangram-suite.readthedocs.io/en/latest/install.html#install-only-one.... Make sure that PLIP does not install numpy, because it will be incompatible with Chimera's. To sum up: 1. Install Miniconda for Py2.7 if you haven't. https://conda.io/miniconda.html 2. Create a new environment: conda create -n plip -c openbabel python=2.7 lxml openbabel 3. Activate the environment: conda activate plip 4. Install PLIP with no deps: pip install pip install https://github.com/ssalentin/plip/archive/master.zip --no-deps # so it does not try to install numpy and openbabel again 5. Install PLIPGUI: pip install future https://github.com/insilichem/libtangram/archive/master.zip https://github.com/insilichem/tangram_plipgui/archive/master.zip 6. Obtain extensions path: echo ${CONDA_PREFIX}/lib/python2.7/site-packages 7. Copy that path and paste it in Chimera> Favorites> Add to Favorites/Tools> Locations. Click Save. A new InsiliChem submenu will be available under Tools. Documentation will be updated here (https://tangram-suite.readthedocs.io/en/latest/tangram_plip.html) at some point, but the usage is straight-forward once it is installed. Just go to Tools> Insilichem> Tangram PLIP, select your opened compound, and voilà. I reckon the install process is not very intuitive because of the added dependencies, so in some future I might port PLIP to be Chimera only (no openbabel deps), thus simplifying the process. Let me know if you need something else. Cheers, Jaime. El vie., 9 nov. 2018 a las 7:57, Francois Berenger (<mlists@ligand.eu<mailto:mlists@ligand.eu>>) escribió: Hello, Recently, I stumbled upon this open source tool: PLIP - Protein-Ligand Interaction Profiler Analyze and visualize non-covalent protein-ligand interactions in PDB files From the abstract: --- [...] detection and visualization of [...] seven interaction types (hydrogen bonds, hydrophobic contacts, pi-stacking, pi-cation interactions, salt bridges, water bridges and halogen bonds). --- Website: http://plip.biotec.tu-dresden.de Open-source code: https://github.com/ssalentin/plip Paper: https://academic.oup.com/nar/article-pdf/43/W1/W443/17435622/gkv315.pdf or https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4489249/ And, when I am inside chimera looking at a protein-ligand complex, it would be super nice if this tool could be launched from within chimera (as in a menu "Tools -> Surface/Binding Analysis -> PLIP analysis (visualize non-covalent interactions)"). Or, maybe it could be a toggable button inside of ViewDock. Best regards, Francois. _______________________________________________ Chimera-users mailing list: Chimera-users@cgl.ucsf.edu<mailto:Chimera-users@cgl.ucsf.edu> Manage subscription: http://plato.cgl.ucsf.edu/mailman/listinfo/chimera-users
{kind=link}
participants (4)
-
 Elaine Meng
Elaine Meng -
 Francois Berenger
Francois Berenger -
 Jaime Rodríguez-Guerra Pedregal
Jaime Rodríguez-Guerra Pedregal -
 zheng l
zheng l