
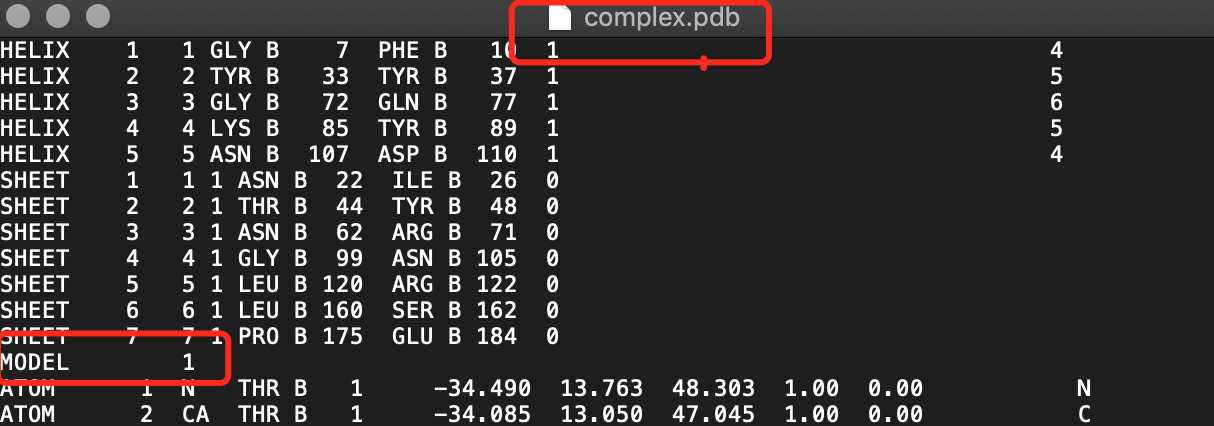
Hello, Dear Chimera developers, I encountered some problems when dealing with complexes of peptide-protein docking,Chimera could not correctly recognize the peptide as a single ligand, but recognized the peptide as model 2 and the receptor protein as model 1. Like this: I would appreciate it if you could help me answer this question. Thank you. Zhuangwei zhang Zhejiang Oeacn University
{kind=link}

Hi Zhuangwei Zhang, Chimera is not doing any "recognizing," this is just because you have the protein and peptide in two different models because they were opened from two different files. This output PDB file has them as two models, which is OK for many programs. However, If you want to write a PDB file where both structures are in one model, you have to first combine them. In Chimera you can combine models with the "copy/combine" function in the Model Panel (open Model Panel from Favorites menu, choose the two models on the left, i.e. highlight both rows with the mouse, then click the "copy/combine" button on the right, OR you can use the "combine" command. copy/combine in Model Panel <https://www.rbvi.ucsf.edu/chimera/docs/UsersGuide/modelpanel.html#combine> combine command: <https://www.rbvi.ucsf.edu/chimera/docs/UsersGuide/midas/combine.html> Then just save the new combined model as a PDB file. I hope this helps, Elaine ----- Elaine C. Meng, Ph.D. UCSF Chimera(X) team Department of Pharmaceutical Chemistry University of California, San Francisco
On Sep 11, 2021, at 8:13 AM, Zhuangwei Zhang via Chimera-users <chimera-users@cgl.ucsf.edu> wrote:
Hello, Dear Chimera developers, I encountered some problems when dealing with complexes of peptide-protein docking,Chimera could not correctly recognize the peptide as a single ligand, but recognized the peptide as model 2 and the receptor protein as model 1. Like this:<781690BA-E872-401A-96D1-4F34E432BFE8.png> I would appreciate it if you could help me answer this question. Thank you. Zhuangwei zhang Zhejiang Oeacn University
<ligand.pdb><receptor.pdb><complex.pdb>x
Dear Elaine C. Meng, Ph.D., Thank you very much for your suggestion. Currently, there are not many people using chimera software in China. This is because the professors are using PyMOL, so the students do not have a good understanding of the ease of use of chimera software. I am translating and promoting the basic manual of chimera software. I look forward to more people knowing and using this software in the future. Thanks again for your help! Zhuangwei zhang Zhejiang Oeacn University On 09/13/2021 01:08,Elaine Meng<meng@cgl.ucsf.edu> wrote: Hi Zhuangwei Zhang, Chimera is not doing any "recognizing," this is just because you have the protein and peptide in two different models because they were opened from two different files. This output PDB file has them as two models, which is OK for many programs. However, If you want to write a PDB file where both structures are in one model, you have to first combine them. In Chimera you can combine models with the "copy/combine" function in the Model Panel (open Model Panel from Favorites menu, choose the two models on the left, i.e. highlight both rows with the mouse, then click the "copy/combine" button on the right, OR you can use the "combine" command. copy/combine in Model Panel <https://www.rbvi.ucsf.edu/chimera/docs/UsersGuide/modelpanel.html#combine> combine command: <https://www.rbvi.ucsf.edu/chimera/docs/UsersGuide/midas/combine.html> Then just save the new combined model as a PDB file. I hope this helps, Elaine ----- Elaine C. Meng, Ph.D. UCSF Chimera(X) team Department of Pharmaceutical Chemistry University of California, San Francisco On Sep 11, 2021, at 8:13 AM, Zhuangwei Zhang via Chimera-users <chimera-users@cgl.ucsf.edu> wrote: Hello, Dear Chimera developers, I encountered some problems when dealing with complexes of peptide-protein docking,Chimera could not correctly recognize the peptide as a single ligand, but recognized the peptide as model 2 and the receptor protein as model 1. Like this:<781690BA-E872-401A-96D1-4F34E432BFE8.png> I would appreciate it if you could help me answer this question. Thank you. Zhuangwei zhang Zhejiang Oeacn University <ligand.pdb><receptor.pdb><complex.pdb>x
Thanks for helping people use Chimera! I encourage you to take a look at ChimeraX, our newer program. If we make new tools and developments, they will be in ChimeraX (not Chimera), and it already has many improvements relative to Chimera. It also has many of the most popular features of Chimera, and I also think it is easier to use. <https://www.rbvi.ucsf.edu/chimerax/index.html> <https://www.rbvi.ucsf.edu/chimerax/docs/user/advantages.html> <https://www.rbvi.ucsf.edu/chimerax/docs/user/index.html> Best regards, Elaine ----- Elaine C. Meng, Ph.D. UCSF Chimera(X) team Department of Pharmaceutical Chemistry University of California, San Francisco
On Sep 12, 2021, at 6:30 PM, Zhuangwei Zhang via Chimera-users <chimera-users@cgl.ucsf.edu> wrote:
Dear Elaine C. Meng, Ph.D., Thank you very much for your suggestion. Currently, there are not many people using chimera software in China. This is because the professors are using PyMOL, so the students do not have a good understanding of the ease of use of chimera software. I am translating and promoting the basic manual of chimera software. I look forward to more people knowing and using this software in the future. Thanks again for your help! Zhuangwei zhang Zhejiang Oeacn University
On 09/13/2021 01:08,Elaine Meng<meng@cgl.ucsf.edu> wrote: Hi Zhuangwei Zhang, Chimera is not doing any "recognizing," this is just because you have the protein and peptide in two different models because they were opened from two different files. This output PDB file has them as two models, which is OK for many programs. However, If you want to write a PDB file where both structures are in one model, you have to first combine them.
In Chimera you can combine models with the "copy/combine" function in the Model Panel (open Model Panel from Favorites menu, choose the two models on the left, i.e. highlight both rows with the mouse, then click the "copy/combine" button on the right, OR you can use the "combine" command.
copy/combine in Model Panel <https://www.rbvi.ucsf.edu/chimera/docs/UsersGuide/modelpanel.html#combine>
combine command: <https://www.rbvi.ucsf.edu/chimera/docs/UsersGuide/midas/combine.html>
Then just save the new combined model as a PDB file.
I hope this helps, Elaine ----- Elaine C. Meng, Ph.D. UCSF Chimera(X) team Department of Pharmaceutical Chemistry University of California, San Francisco
On Sep 11, 2021, at 8:13 AM, Zhuangwei Zhang via Chimera-users <chimera-users@cgl.ucsf.edu> wrote:
Hello, Dear Chimera developers, I encountered some problems when dealing with complexes of peptide-protein docking,Chimera could not correctly recognize the peptide as a single ligand, but recognized the peptide as model 2 and the receptor protein as model 1. Like this:<781690BA-E872-401A-96D1-4F34E432BFE8.png> I would appreciate it if you could help me answer this question. Thank you. Zhuangwei zhang Zhejiang Oeacn University
<ligand.pdb><receptor.pdb><complex.pdb>x _______________________________________________ Chimera-users mailing list: Chimera-users@cgl.ucsf.edu Manage subscription: https://plato.cgl.ucsf.edu/mailman/listinfo/chimera-users
participants (2)
-
 Elaine Meng
Elaine Meng -
 Zhuangwei Zhang
Zhuangwei Zhang