
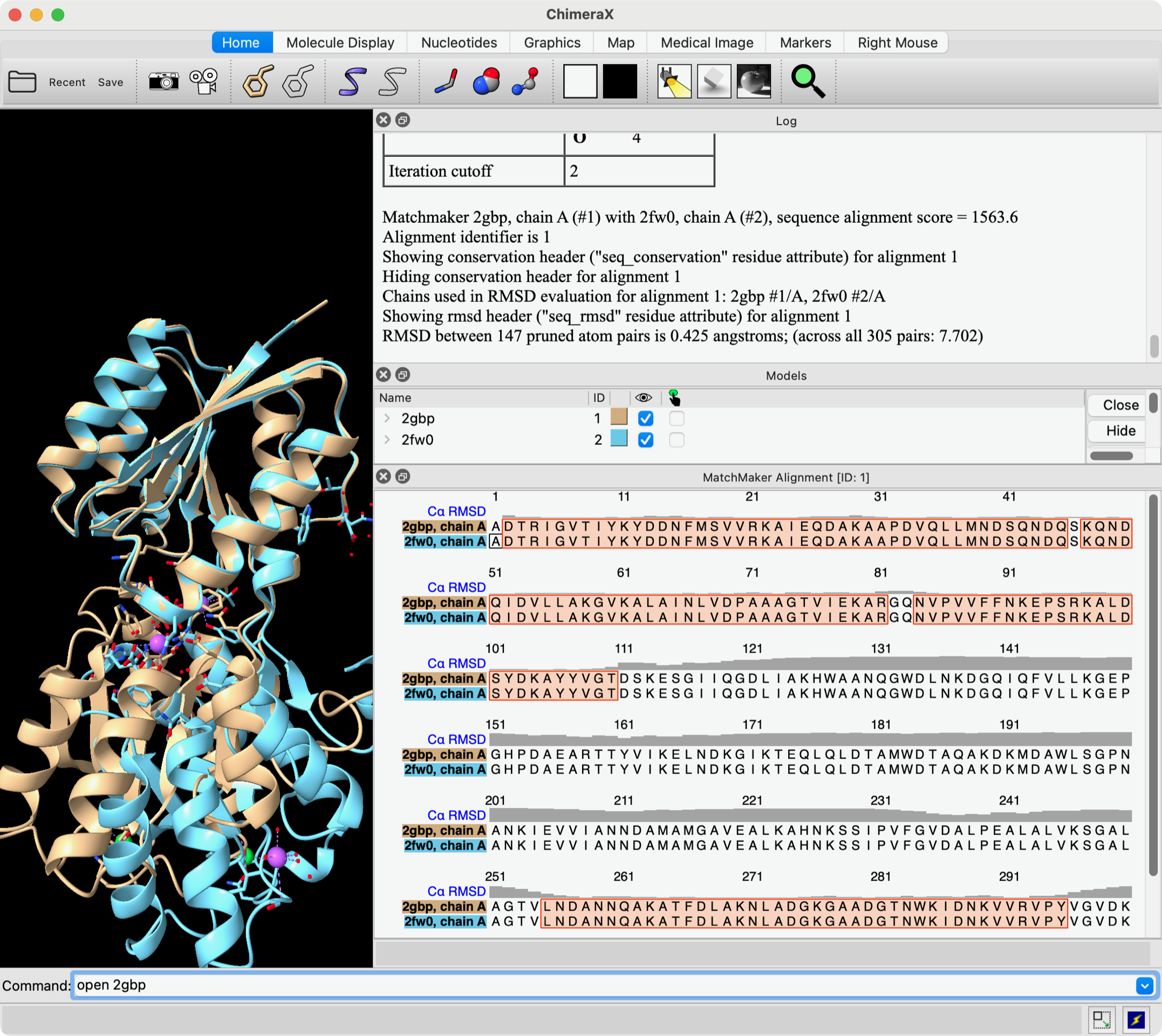
Hi Diego, As far as I know there is no measurement (in any program, I mean) that is the % similarity in structures. There are lots of other structure similarity measures. What people use the most is probably RMSD and that is already reported in the Log after you use matchmaker. Matchmaker uses only one atom per residue, CA atoms in amino acids, to calculate this RMSD. For example, these commands: open 2gbp open 2fw0 mm #2 to #1 show true ... open two structures and run Matchmaker, and result is shown in this screenshot. The "show true" shows the sequence alignment and the positions used for the final fit are outlined in orange boxes on the alignment. The Log window shows that the RMSD for the 147 positions (CA-CA pairs) in the orange boxes is 0.425 angstroms, and also across the entire sequences (305 CA-CA) pairs is 7.702 angstroms. The gray histogram over the sequences shows the CA-CA distance at each position. If you want to use more atoms (not just one CA atom per residue) to calculate the RMSD without moving the structures from their current positions, then you could use the "rmsd" command. See: <https://rbvi.ucsf.edu/chimerax/docs/user/commands/rmsd.html> Elaine ----- Elaine C. Meng, Ph.D. UCSF Chimera(X) team Resource for Biocomputing, Visualization, and Informatics Department of Pharmaceutical Chemistry University of California, San Francisco
On Jul 2, 2024, at 2:12 AM, DIEGO ANGOSTO BAZARRA via ChimeraX-users <chimerax-users@cgl.ucsf.edu> wrote:
Hello,
My name is Diego. I’m a beginner user of ChimeraX (1.8) and I was wondering if there is an option to check the % of similarity between two structures after making the matchmaker.
I mean, I make a matchmaker with two structures, can I see the % of similarity between these two structures? Not between the sequences, but between the structures.
Thank you very much for your time and attention.
Best regards
Dr. Diego Angosto Bazarra Molecular inflammation and experimental surgery group. IMIB. University Hospital Virgen de la Arrixaca
{kind=link}