
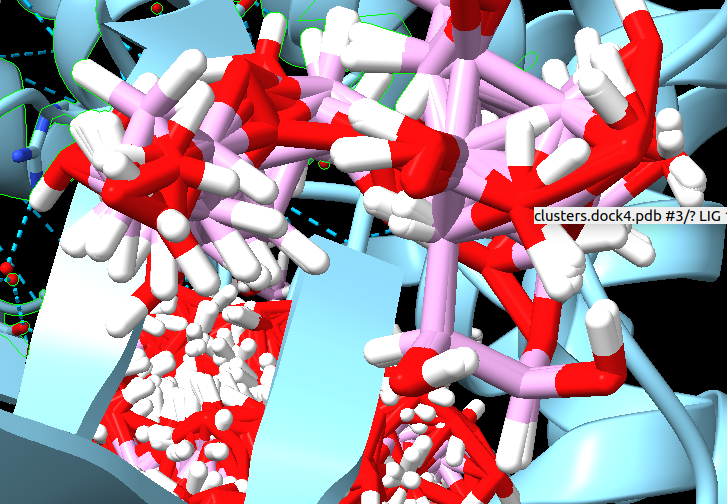
Dear sir or madam, I am new to docking analysis. I obtained a model from SwissDock but there are some commands that do not work on ChimeraX. 1. I can load the molecule, which is composed by 4 homo-chains. I can select one chain (A); how can I delete or hide chains B-D. I can select chain A using select>chain`; is there a way to hide the others? 2. The model (saved in the file clusters.dock4.pdb) shows several ligands: [cid:c289e47f-ca49-4f27-9ad6-acef069f976e][cid:728d8f47-8cfb-445c-9476-ebb8a9d54ef9] Is there a way to show only the ligands with the highest docking score? 3. the tools>binding analysis>ViewDockX` says that "No suitable models found for ViewDockX". But is not this the "clusters.dock4.pdb"? How can I load the model into ChimeraX? Thank you. Best regards, Luigi Marongiu Ph.D., B.Sc. (Hons.)
{kind=link}
{kind=link}