
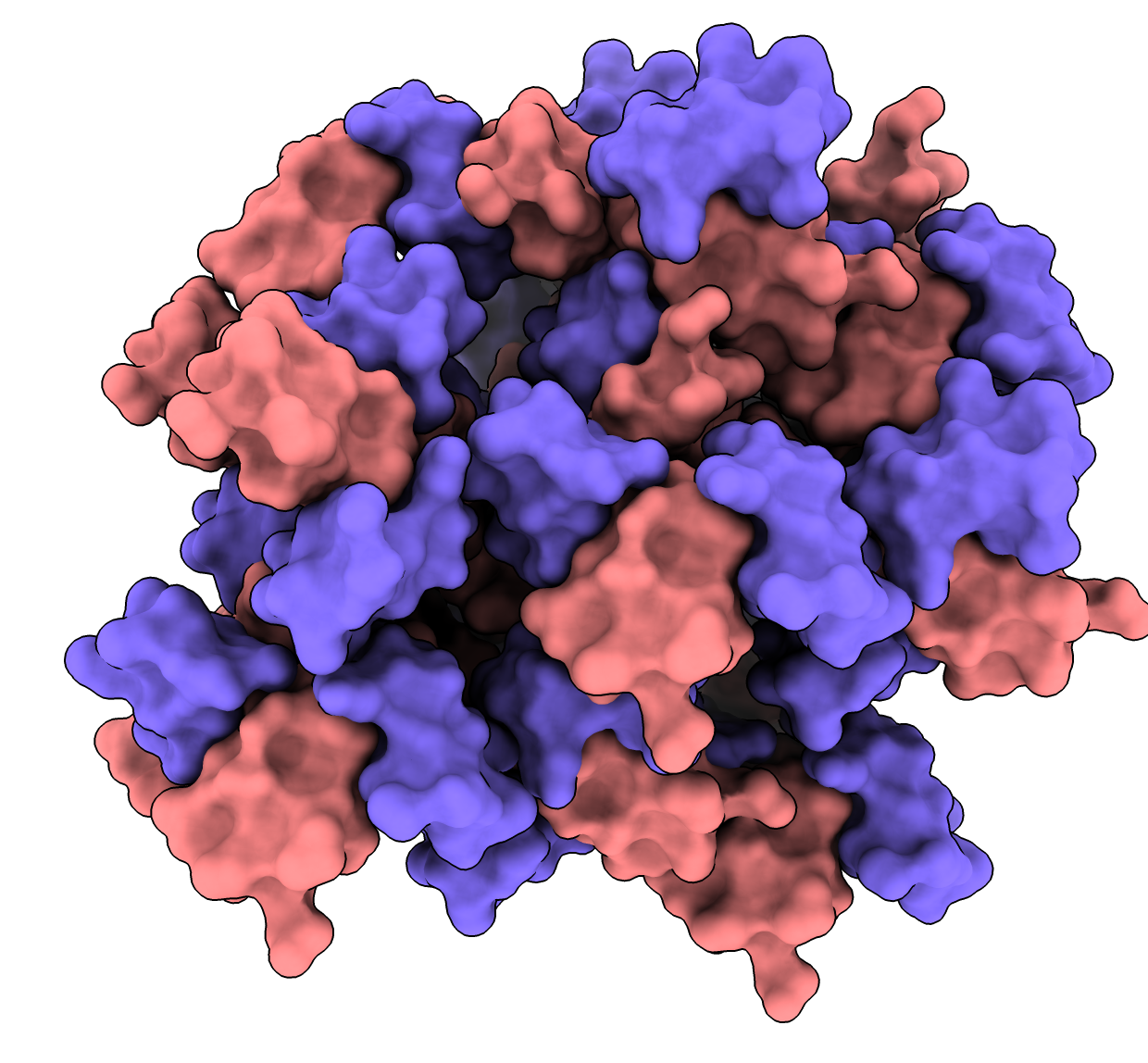
Hi Guillaume, Showing contacting asymmetric units in crystal hasn't been added to ChimeraX, but I will add it. What you want is the crystalcontacts command from Chimera. https://www.cgl.ucsf.edu/chimera/current/docs/UsersGuide/midas/crystalcontac... Chimera has 25 years of accumulated features and we are moving all the good stuff to ChimeraX but it takes time. During the transition I advise using both programs for what they do best. I would make the crystal contacts in Chimera, write a PDB, and open it in ChimeraX. Here's an example. In Chimera, open 1a0m crystalcontacts #0 10 copies true schematic false menu File / Save PDB..., save all in a single PDB file 1a0m_range10.pdb In ChimeraX open 1a0m_range10.pdb show surface Tom
On May 8, 2020, at 6:24 AM, Guillaume Gaullier <guillaume@gaullier.org> wrote:
Hello,
Today I was trying to display a crystal packing from a PDB entry in ChimeraX, but could not find how to do this. I read the documentation for the sym command https://www.cgl.ucsf.edu/chimerax/docs/user/commands/sym.html ; but either I didn’t get it, or this is not the adequate command for what I am trying to do.
What I want to do is the equivalent of PyMOL’s "Action / generate / symmetry mates / +/- one unit cell and within… Å", which is different from the biological assembly the sym command can generate from the PDB annotation. When working with crystallographic data, ISOLDE displays molecules related to the working model by crystallographic symmetry, so displaying the crystal packing seems doable in ChimeraX. Or is it one of ISOLDE’s additions?
It would be wonderful if someone could help me with this (it is one of the last few things I need on a regular basis, that I still don’t know how to do in ChimeraX).
Thank you in advance,
Guillaume
_______________________________________________ ChimeraX-users mailing list ChimeraX-users@cgl.ucsf.edu Manage subscription: http://www.rbvi.ucsf.edu/mailman/listinfo/chimerax-users
{kind=link}