
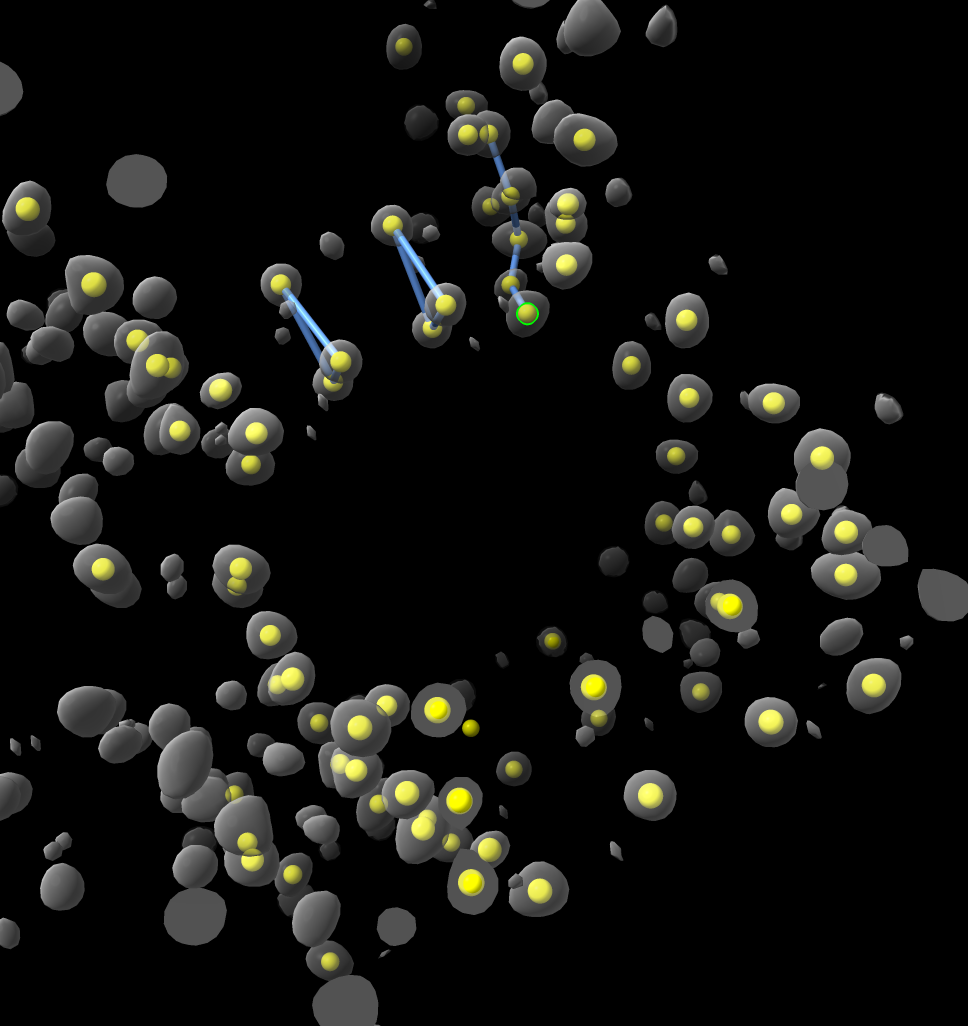
Hi Wang, I think as Elaine suggests just placing markers on the density map might get you started. I tried it using the ChimeraX Markers toolbar using place markers on Center mouse mode. Then just right-clicked 50 blobs to place markers, then used the Link markers mouse mode to connect a few, then wrote out an mmCIF file. All seemed doable. Of course the atom types, silicon vs oxygen are not distinguished but you could perhaps hand edit the mmCIF file to fix that. Tom
On Jul 28, 2020, at 1:23 AM, 王超 <wangchao17@mails.jlu.edu.cn> wrote:
Dear professor I found ChimeraX software from the Internet. Through interface design, I think this should be a very powerful software. Now, I have a electron density map of zeolite in ''.xplor'' format. By adjusting the threshold, I can clearly see where the atom is. So I wonder whether it is possible to use this software to export the coordinates of the structural atoms,especially ''.cif '' format files. Thank you for your help !
-- Best Wishes!
Wang, Chao Jilin University 2699 Qianjin Street, Changchun 130012, China Email:wangchao17@mails.jlu.edu.cn <mailto:wangchao17@mails.jlu.edu.cn> <IFT.png><IFT.xplor>_______________________________________________ ChimeraX-users mailing list ChimeraX-users@cgl.ucsf.edu Manage subscription: https://www.rbvi.ucsf.edu/mailman/listinfo/chimerax-users
{kind=link}