
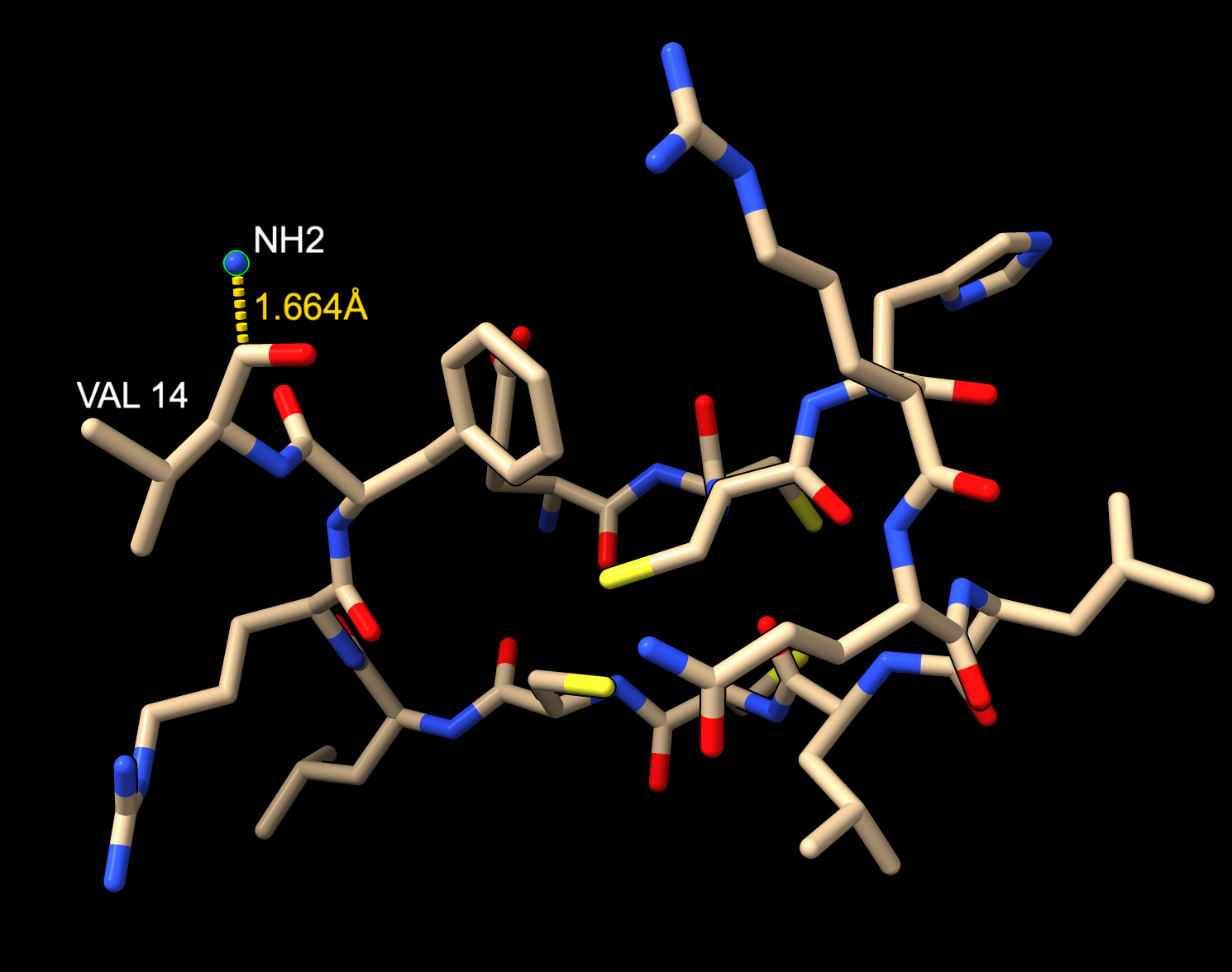
Hi Ryssa, It is possible to use Boltz with ChimeraX to predict an amidated peptide in complex with a protein. As Elaine pointed out the ChimeraX user interface doesn't have a way to do the amidation. But you can run the ChimeraX Boltz tool without the amidation (menu Tools / Structure Prediction / Boltz), and then edit the Boltz input file ChimeraX created to add the amidation and rerun Boltz from a terminal with that new input file. I tried it amidating a 14 amino acid peptide with a valine at the C-terminus. Unfortunately Boltz did not make a good prediction. The added amide was not shown as bonded to the valine, probably that is just a problem with the mmCIF output file, but more troubling the distance to the added amide was too long (1.7 Angstroms), image below. I don't think this is going to be a promising solution for you. But for completeness here are details of what I tried. I used the sequence from amidated-petide structure PDB 3zkt, ECCHRQLLCCLRFV. ChimeraX ran a prediction of this using the following Boltz input file that it created version: 1 sequences: - protein: id: [A] sequence: ECCHRQLLCCLRFV I copied that Boltz input file 3kzt.yaml to a new file 3kzt_nh2.yaml and added the amidation as follows (make sure to use spaces not tabs). version: 1 sequences: - protein: id: [A] sequence: ECCHRQLLCCLRFV - ligand: id: [B] ccd: NH2 constraints: - bond: atom1: [A, 14, C] atom2: [B, 1, N] The amide is added as an NH2 ligand, chain B, and then is connected by a bond to the peptide chain A residue 14, C atom. Then I ran a Boltz prediction with this file from a terminal on my Mac using a modified copy of the command ChimeraX used (found in the file ChimeraX created named "command") $ /Users/goddard/boltz22/bin/boltz predict /Users/goddard/Desktop/boltz/boltz_3zkt/3zkt_nh2.yaml --use_msa_server --accelerator gpu --no_kernels --use_potentials >& output_nh2 I added the option --use_potentials which uses Boltz steering potentials to improve geometry. Without that the amide was 2A away. With steering it got a bit closer at 1.7 A. But the 3zkt PDB has it at 1.3 A. It took about 1 minute. I looked at the Boltz prediction by opening this mmCIF file in ChimeraX ~/Desktop/boltz/boltz_3zkt/boltz_results_3zkt_nh2/predictions/3zkt_nh2/3zkt_nh2_model_0.cif and that is shown below. Tom 14 amino acid peptide with amidated C-terminus predicted by Boltz.
On Sep 16, 2025, at 11:49 AM, Elaine Meng via ChimeraX-users <chimerax-users@cgl.ucsf.edu> wrote:
Hi Ryssa, The ChimeraX alphafold tool runs ColabFold, which is based on AlphaFold 2 and does not predict post-translational modifications. <https://rbvi.ucsf.edu/chimerax/docs/user/tools/alphafold.html>
There is another ChimeraX tool that runs Boltz, which is somewhat based on AlphaFold 3. Although the Boltz program can predict post-translational modifications, however, it is a current limitation of the ChimeraX interface (tool and command) that there is no way to specify such modifications. Sorry about that. See <https://rbvi.ucsf.edu/chimerax/docs/user/tools/boltz.html> <https://rbvi.ucsf.edu/chimerax/docs/user/tools/boltz.html#limitations>
I hope this clarifies the current situation, Elaine ----- Elaine C. Meng, Ph.D. UCSF Chimera(X) team Resource for Biocomputing, Visualization, and Informatics Department of Pharmaceutical Chemistry University of California, San Francisco
On Sep 16, 2025, at 11:16 AM, Parks, Ryssa via ChimeraX-users <chimerax-users@cgl.ucsf.edu> wrote:
Hello, I am trying to predict and model a peptide docking to a receptor protein using ChimeraX's AlphaFold interface, and I was wondering if there is any way to specify post-translational modifications in the AlphaFold input function in ChimeraX? Our peptide has an amidated C-terminus, and my searches online have not yielded much success.
Thank you, Ryssa Parks
_______________________________________________ ChimeraX-users mailing list -- chimerax-users@cgl.ucsf.edu To unsubscribe send an email to chimerax-users-leave@cgl.ucsf.edu Archives: https://mail.cgl.ucsf.edu/mailman/archives/list/chimerax-users@cgl.ucsf.edu/
{kind=link}