
Thanks Elaine, this is very helpful. I played around with what you sent me and although it works great, I discovered two little issues with Chimera X: (1) this is really minor, but I love the "soft lighting" setting and wanted to have that be the default setting in my installation. I figured I could set "lighting soft" to execute at startup, but for me, loading a new pdb always changes the lighting back to "simple". After I change it back to soft lighting it will stay that way when I bring in new pdb files without closing the original file, but if for instance I type "close all; open 2ZTA", it's back to simple lighting again. Like I said, a minor issue. But in good ol' Chimera I appreciated that I could set it up just the way I like it. (2) In the color actions GUI, the radio buttons for applying color aren't being incorporated into the command. For example, if I uncheck everything except atoms/bonds, and then click "color by heteroatom", it runs "color byhetero", which applies to all. I had been using 1.3, but I updated to the latest daily build and still have the same issues. Dan ____________________________ Daniel Gurnon, Ph. D. Associate Professor of Chemistry and Biochemistry DePauw University Greencastle, IN 46135 On Mon, Aug 29, 2022 at 2:38 PM Elaine Meng <meng@cgl.ucsf.edu> wrote:
Hi Dan, Thanks for using Chimera & ChimeraX in your teaching!
We do plan to have a Render by Attribute tool in ChimeraX, at least for the coloring part. However, it's a rather complicated GUI that we are still working on implementing from scratch (since ChimeraX uses different toolkits than Chimera).
ChimeraX does already have the command implementation, "color byattribute" <https://rbvi.ucsf.edu/chimerax/docs/user/commands/color.html#byattribute>
Although kdHydrophobicity of amino acid residues is not automatically assigned as an attribute in ChimeraX, we provide:
(1) a file for loading it as an attribute, e.g. download this file as plain text and then use command "open kdHydrophobicity.defattr" <https://rbvi.ucsf.edu/chimerax/docs/user/formats/kdHydrophobicity.defattr
...after which you could color by attribute with the command, or select residues based on their attribute values, label by attribute, etc. There are examples specifically for kdHydrophobicity here: <https://rbvi.ucsf.edu/chimerax/docs/user/formats/defattr.html>
For example, to use the same coloring scheme as in Chimera, command:
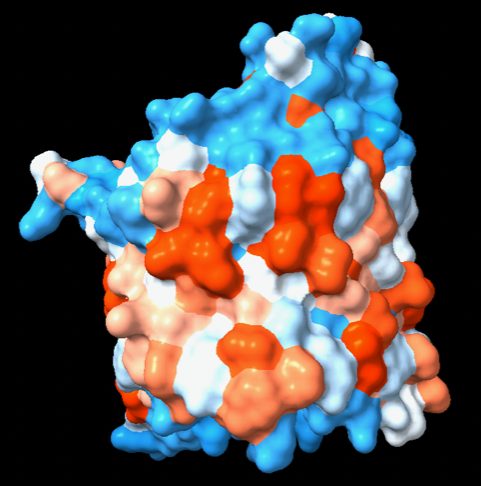
color byattribute kdHydrophobicity protein palette dodgerblue:white:orangered novalue gray
There are also files for other hydrophobicity scales, in case you're interested... <https://rbvi.ucsf.edu/chimerax/docs/user/formats/defattr.html#examples>
(2) A ChimeraX command file (.cxc) that assigns the attribute as name "kdh" (could be changed to kdHydrophobicity if you prefer), shows the protein molecular surface, and colors it by those values using the same color scheme that was used in Chimera. The result looks somewhat different in ChimeraX than Chimera, however, since there are sharper boundaries in the molecular surface between the residues. < https://rbvi.ucsf.edu/chimerax/docs/user/kyte-doolittle_hydrophobicity.cxc
How to use the .cxc file to define a custom preset (i.e. will make an entry in the Presets menu): <https://rbvi.ucsf.edu/chimerax/docs/user/preferences.html#startup>
This .cxc file is just a starting point if you want to do something slightly different. You could remove the commands to hide protein atoms and show surface, you could change the coloring target to include atoms, etc.
I realize these are more steps in than in Chimera, however. If you are in control of the installation that all the students use, it would work well for you to make a custom preset. However, if they are all using their own installations on their own laptops, you could just tell the students to get the .cxc file (as plain text) and simply run the script by opening it in ChimeraX every time they want to do the coloring.
I hope this helps, Elaine ----- Elaine C. Meng, Ph.D. UCSF Chimera(X) team Department of Pharmaceutical Chemistry University of California, San Francisco
On Aug 29, 2022, at 9:20 AM, Daniel Gurnon via ChimeraX-users < chimerax-users@cgl.ucsf.edu> wrote:
Hi all, I use Chimera mostly for teaching purposes, and while Chimera X is definitely more user friendly for students, there are occasionally things I like from the legacy version that I can't find on a menu in the new program. One such feature is "Render by attribute". While the mlp command can create a lipophilicity surface, when I teach protein folding I like to have students visualize all atoms, with residues colored by kdHydrophobicity. Is there a simple way to implement this?
Thanks Dan ____________________________
Daniel Gurnon, Ph. D. Associate Professor of Chemistry and Biochemistry DePauw University Greencastle, IN 46135 _______________________________________________ ChimeraX-users mailing list ChimeraX-users@cgl.ucsf.edu Manage subscription: https://www.rbvi.ucsf.edu/mailman/listinfo/chimerax-users
{kind=link}
{kind=link}