Reading cube file from Quantum Espresso pp.x

Hi, I produced some projected wavefunctions in a crystal that I'm working with using Quantum Espresso (DFT). I output these projections to a gaussian cube file using the pp.x program (built into Quantum Espresso). I'm able to read these into ChimeraX but I'm unable to see the atoms -- only the volumetric data. I tried Actions - Atoms/Bonds - Show, but I still don't see the atoms. Is there another step I should take to make them visible? I see something like this: (very nice!) I'm attaching a link to the cube file below -- I can't include it because it is 600 MB. Thanks, Zack https://www.dropbox.com/s/ehhli8h3qr7vd4y/wfc_k001_b329wfc.cube?dl=0
{kind=link}

Hi Zack, A cube file is a map format (values at points on a regular grid), and is not used for atomic coordinates. In other words, I believe your file contains only the wavefunctions. You would need to also open a file for the atomic coordinates (possible formats PDB, SDF, MOL, ...) which you may have input to Quantum Espresso, or may be able to write from the program. I've never used it, so I don't know what it writes. ChimeraX knows these atomic formats: <https://rbvi.ucsf.edu/chimerax/docs/user/commands/open.html#atomic> ... and these map formats: <https://rbvi.ucsf.edu/chimerax/docs/user/commands/open.html#volume> I hope this helps, Elaine ----- Elaine C. Meng, Ph.D. UCSF Chimera(X) team Department of Pharmaceutical Chemistry University of California, San Francisco
On Feb 2, 2021, at 9:45 AM, Zack Gainsforth <zackg@berkeley.edu> wrote:
Hi,
I produced some projected wavefunctions in a crystal that I'm working with using Quantum Espresso (DFT). I output these projections to a gaussian cube file using the pp.x program (built into Quantum Espresso). I'm able to read these into ChimeraX but I'm unable to see the atoms -- only the volumetric data. I tried Actions - Atoms/Bonds - Show, but I still don't see the atoms. Is there another step I should take to make them visible?
I see something like this:
<PastedGraphic-1.png>
(very nice!)
I'm attaching a link to the cube file below -- I can't include it because it is 600 MB.
Thanks,
Zack
https://www.dropbox.com/s/ehhli8h3qr7vd4y/wfc_k001_b329wfc.cube?dl=0
Hi Elaine, Thanks for the feedback! I think it does contain atomic coordinates because if I open the file up, the first few lines say: Cubefile created from PWScf calculation k_point 1, band 493 112 0.000000 0.000000 0.000000 360 0.000000 0.043010 0.043010 360 0.043010 0.000000 0.043010 360 0.043010 0.043010 0.000000 13 13.000000 15.550626 7.919837 23.249520 13 13.000000 7.794637 15.500714 23.173025 13 13.000000 15.344622 7.808598 7.696103 24 24.000000 8.153563 23.302893 15.676357 24 24.000000 22.842015 15.147909 23.267895 24 24.000000 23.276582 23.168925 15.892733 24 24.000000 0.413132 15.595221 15.687712 13 13.000000 15.337306 19.401078 19.211118 24 24.000000 23.238956 26.684104 19.354277 13 13.000000 30.836280 19.498740 19.483616 13 13.000000 23.156913 11.704068 19.344564 13 13.000000 7.845982 3.847472 11.610165 So we have 13 being magnesium, and 24 being Cr, etc. After the 112 atoms, it starts with the volumetric values. So it sounds like perhaps I should request a feature addition to ChimeraX to implement atomic coordinates as well? Following your advice though, I saved the structure as a pdb also, and loaded that and it seems to work — though there is an offset between the volumetric and atomic datasets. I guess I would need to manually shift one of them, so that provides a workaround for now. Thanks, Zack
On Feb 2, 2021, at 10:15 AM, Elaine Meng <meng@cgl.ucsf.edu> wrote:
Hi Zack, A cube file is a map format (values at points on a regular grid), and is not used for atomic coordinates. In other words, I believe your file contains only the wavefunctions. You would need to also open a file for the atomic coordinates (possible formats PDB, SDF, MOL, ...) which you may have input to Quantum Espresso, or may be able to write from the program. I've never used it, so I don't know what it writes.
ChimeraX knows these atomic formats: <https://rbvi.ucsf.edu/chimerax/docs/user/commands/open.html#atomic> ... and these map formats: <https://rbvi.ucsf.edu/chimerax/docs/user/commands/open.html#volume>
I hope this helps, Elaine ----- Elaine C. Meng, Ph.D. UCSF Chimera(X) team Department of Pharmaceutical Chemistry University of California, San Francisco
On Feb 2, 2021, at 9:45 AM, Zack Gainsforth <zackg@berkeley.edu> wrote:
Hi,
I produced some projected wavefunctions in a crystal that I'm working with using Quantum Espresso (DFT). I output these projections to a gaussian cube file using the pp.x program (built into Quantum Espresso). I'm able to read these into ChimeraX but I'm unable to see the atoms -- only the volumetric data. I tried Actions - Atoms/Bonds - Show, but I still don't see the atoms. Is there another step I should take to make them visible?
I see something like this:
<PastedGraphic-1.png>
(very nice!)
I'm attaching a link to the cube file below -- I can't include it because it is 600 MB.
Thanks,
Zack
https://www.dropbox.com/s/ehhli8h3qr7vd4y/wfc_k001_b329wfc.cube?dl=0
{kind=link}

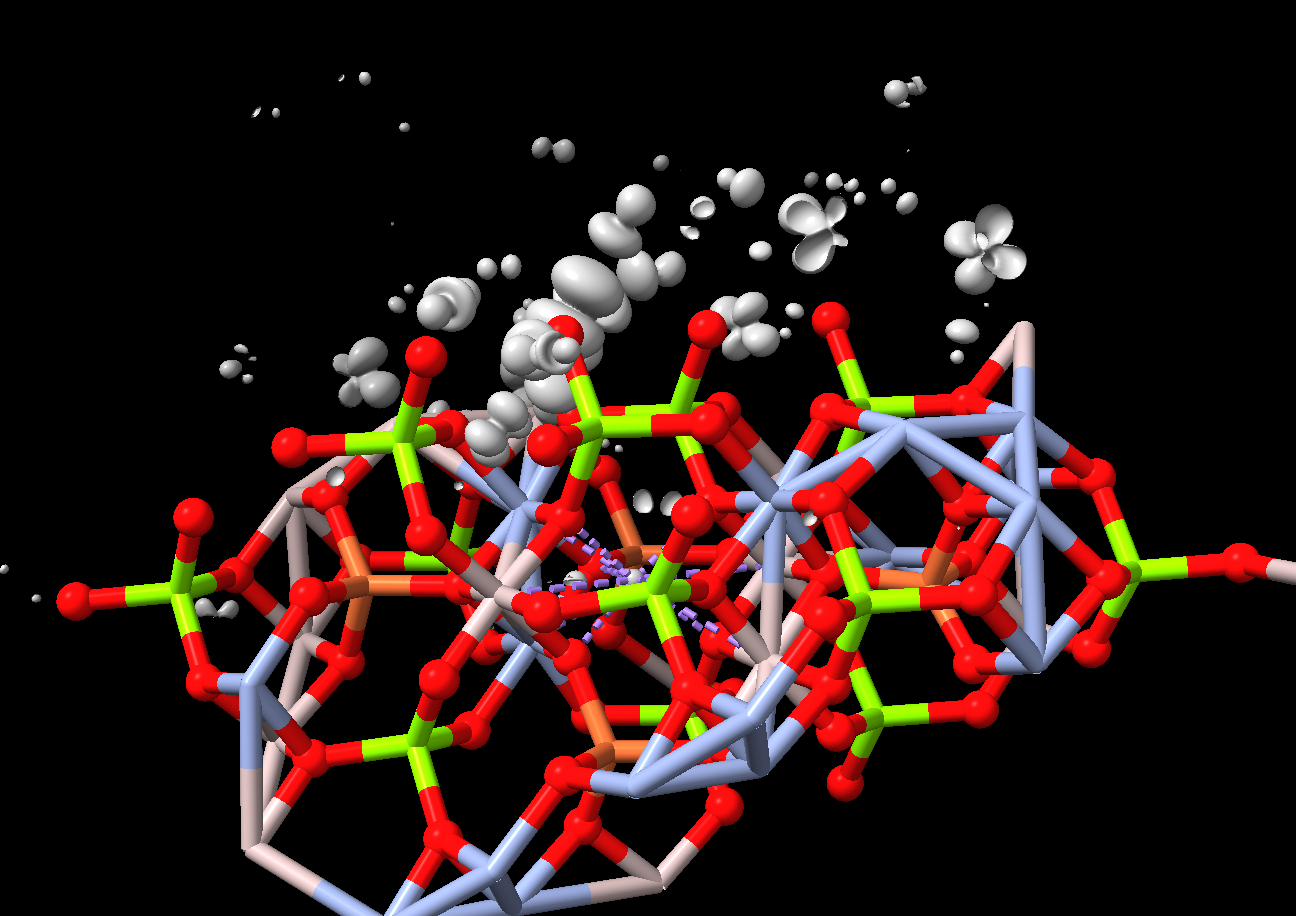
Hi Zack, Elaine is right that ChimeraX only reads the map in Gaussian cube files, not the atoms. I was curious what your atoms looked like so I wrote a short Python script to make the atoms from your file and put it on the ChimeraX Recipes web site as an example of ChimeraX Python code. https://rbvi.github.io/chimerax-recipes/gaussian_cube_atoms/gcatoms.html The Gaussian cube file you linked to in your original message is not the same as the one you show in your second message. In the linked file I see a lattice of aluminum, magnesium and oxygen. ChimeraX tries to make metal coordination bonds (purple dashed lines) and put bonds between various aluminum atoms and made a pretty bizarre and likely wrong picture. ChimeraX is designed for proteins so doesn't know what to do with inorganic crystals. Tom
On Feb 2, 2021, at 10:42 AM, Zack Gainsforth <zackg@berkeley.edu> wrote:
Hi Elaine,
Thanks for the feedback!
I think it does contain atomic coordinates because if I open the file up, the first few lines say:
Cubefile created from PWScf calculation k_point 1, band 493 112 0.000000 0.000000 0.000000 360 0.000000 0.043010 0.043010 360 0.043010 0.000000 0.043010 360 0.043010 0.043010 0.000000 13 13.000000 15.550626 7.919837 23.249520 13 13.000000 7.794637 15.500714 23.173025 13 13.000000 15.344622 7.808598 7.696103 24 24.000000 8.153563 23.302893 15.676357 24 24.000000 22.842015 15.147909 23.267895 24 24.000000 23.276582 23.168925 15.892733 24 24.000000 0.413132 15.595221 15.687712 13 13.000000 15.337306 19.401078 19.211118 24 24.000000 23.238956 26.684104 19.354277 13 13.000000 30.836280 19.498740 19.483616 13 13.000000 23.156913 11.704068 19.344564 13 13.000000 7.845982 3.847472 11.610165
So we have 13 being magnesium, and 24 being Cr, etc.
After the 112 atoms, it starts with the volumetric values.
So it sounds like perhaps I should request a feature addition to ChimeraX to implement atomic coordinates as well?
Following your advice though, I saved the structure as a pdb also, and loaded that and it seems to work — though there is an offset between the volumetric and atomic datasets.
<PastedGraphic-1.png>
I guess I would need to manually shift one of them, so that provides a workaround for now.
Thanks,
Zack
On Feb 2, 2021, at 10:15 AM, Elaine Meng <meng@cgl.ucsf.edu <mailto:meng@cgl.ucsf.edu>> wrote:
Hi Zack, A cube file is a map format (values at points on a regular grid), and is not used for atomic coordinates. In other words, I believe your file contains only the wavefunctions. You would need to also open a file for the atomic coordinates (possible formats PDB, SDF, MOL, ...) which you may have input to Quantum Espresso, or may be able to write from the program. I've never used it, so I don't know what it writes.
ChimeraX knows these atomic formats: <https://rbvi.ucsf.edu/chimerax/docs/user/commands/open.html#atomic <https://rbvi.ucsf.edu/chimerax/docs/user/commands/open.html#atomic>> ... and these map formats: <https://rbvi.ucsf.edu/chimerax/docs/user/commands/open.html#volume <https://rbvi.ucsf.edu/chimerax/docs/user/commands/open.html#volume>>
I hope this helps, Elaine ----- Elaine C. Meng, Ph.D. UCSF Chimera(X) team Department of Pharmaceutical Chemistry University of California, San Francisco
On Feb 2, 2021, at 9:45 AM, Zack Gainsforth <zackg@berkeley.edu <mailto:zackg@berkeley.edu>> wrote:
Hi,
I produced some projected wavefunctions in a crystal that I'm working with using Quantum Espresso (DFT). I output these projections to a gaussian cube file using the pp.x program (built into Quantum Espresso). I'm able to read these into ChimeraX but I'm unable to see the atoms -- only the volumetric data. I tried Actions - Atoms/Bonds - Show, but I still don't see the atoms. Is there another step I should take to make them visible?
I see something like this:
<PastedGraphic-1.png>
(very nice!)
I'm attaching a link to the cube file below -- I can't include it because it is 600 MB.
Thanks,
Zack
https://www.dropbox.com/s/ehhli8h3qr7vd4y/wfc_k001_b329wfc.cube?dl=0 <https://www.dropbox.com/s/ehhli8h3qr7vd4y/wfc_k001_b329wfc.cube?dl=0>
_______________________________________________ ChimeraX-users mailing list ChimeraX-users@cgl.ucsf.edu Manage subscription: https://www.rbvi.ucsf.edu/mailman/listinfo/chimerax-users
{kind=link}
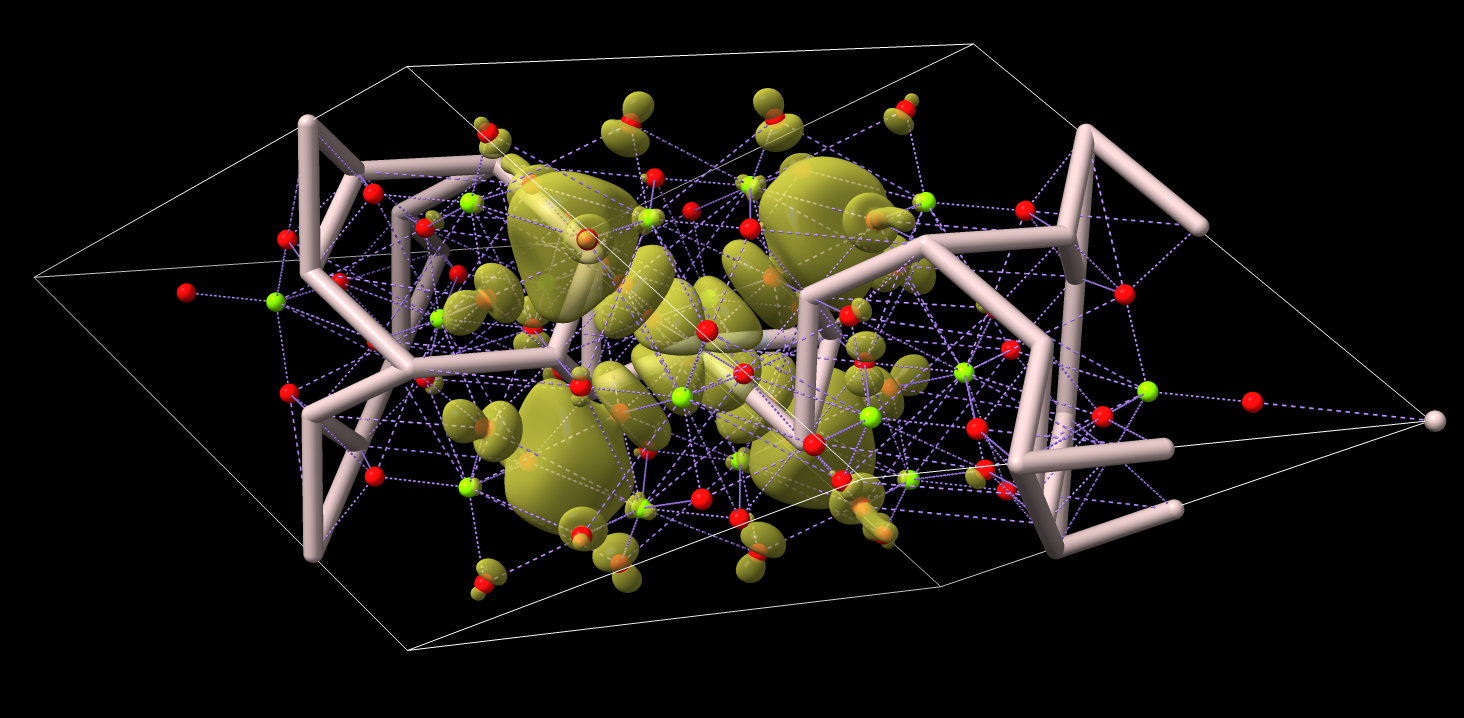
Hi Tom, Sorry if I got my cube files crossed up! I’m comparing several at the present so I must have just mixed it up. The latest image you sent looks correct, except for the bonds as you note. (It’s a spinel.) However, the projected orbitals look correct, so the code is superimposing the volumetric data on the atomic data correctly. I tried it your script and it works terrifically! (Although I think some indentation got lost in the post on github.) I simply turned off the metallic bonds and wound up with (yet another cube file): Thank you much! Cheers, Zack
On Feb 2, 2021, at 11:19 AM, Tom Goddard <goddard@sonic.net> wrote:
Hi Zack,
Elaine is right that ChimeraX only reads the map in Gaussian cube files, not the atoms. I was curious what your atoms looked like so I wrote a short Python script to make the atoms from your file and put it on the ChimeraX Recipes web site as an example of ChimeraX Python code.
https://rbvi.github.io/chimerax-recipes/gaussian_cube_atoms/gcatoms.html <https://rbvi.github.io/chimerax-recipes/gaussian_cube_atoms/gcatoms.html>
The Gaussian cube file you linked to in your original message is not the same as the one you show in your second message. In the linked file I see a lattice of aluminum, magnesium and oxygen. ChimeraX tries to make metal coordination bonds (purple dashed lines) and put bonds between various aluminum atoms and made a pretty bizarre and likely wrong picture. ChimeraX is designed for proteins so doesn't know what to do with inorganic crystals.
Tom
<wfc_K001_B329wfc.png>
On Feb 2, 2021, at 10:42 AM, Zack Gainsforth <zackg@berkeley.edu <mailto:zackg@berkeley.edu>> wrote:
Hi Elaine,
Thanks for the feedback!
I think it does contain atomic coordinates because if I open the file up, the first few lines say:
Cubefile created from PWScf calculation k_point 1, band 493 112 0.000000 0.000000 0.000000 360 0.000000 0.043010 0.043010 360 0.043010 0.000000 0.043010 360 0.043010 0.043010 0.000000 13 13.000000 15.550626 7.919837 23.249520 13 13.000000 7.794637 15.500714 23.173025 13 13.000000 15.344622 7.808598 7.696103 24 24.000000 8.153563 23.302893 15.676357 24 24.000000 22.842015 15.147909 23.267895 24 24.000000 23.276582 23.168925 15.892733 24 24.000000 0.413132 15.595221 15.687712 13 13.000000 15.337306 19.401078 19.211118 24 24.000000 23.238956 26.684104 19.354277 13 13.000000 30.836280 19.498740 19.483616 13 13.000000 23.156913 11.704068 19.344564 13 13.000000 7.845982 3.847472 11.610165
So we have 13 being magnesium, and 24 being Cr, etc.
After the 112 atoms, it starts with the volumetric values.
So it sounds like perhaps I should request a feature addition to ChimeraX to implement atomic coordinates as well?
Following your advice though, I saved the structure as a pdb also, and loaded that and it seems to work — though there is an offset between the volumetric and atomic datasets.
<PastedGraphic-1.png>
I guess I would need to manually shift one of them, so that provides a workaround for now.
Thanks,
Zack
On Feb 2, 2021, at 10:15 AM, Elaine Meng <meng@cgl.ucsf.edu <mailto:meng@cgl.ucsf.edu>> wrote:
Hi Zack, A cube file is a map format (values at points on a regular grid), and is not used for atomic coordinates. In other words, I believe your file contains only the wavefunctions. You would need to also open a file for the atomic coordinates (possible formats PDB, SDF, MOL, ...) which you may have input to Quantum Espresso, or may be able to write from the program. I've never used it, so I don't know what it writes.
ChimeraX knows these atomic formats: <https://rbvi.ucsf.edu/chimerax/docs/user/commands/open.html#atomic <https://rbvi.ucsf.edu/chimerax/docs/user/commands/open.html#atomic>> ... and these map formats: <https://rbvi.ucsf.edu/chimerax/docs/user/commands/open.html#volume <https://rbvi.ucsf.edu/chimerax/docs/user/commands/open.html#volume>>
I hope this helps, Elaine ----- Elaine C. Meng, Ph.D. UCSF Chimera(X) team Department of Pharmaceutical Chemistry University of California, San Francisco
On Feb 2, 2021, at 9:45 AM, Zack Gainsforth <zackg@berkeley.edu <mailto:zackg@berkeley.edu>> wrote:
Hi,
I produced some projected wavefunctions in a crystal that I'm working with using Quantum Espresso (DFT). I output these projections to a gaussian cube file using the pp.x program (built into Quantum Espresso). I'm able to read these into ChimeraX but I'm unable to see the atoms -- only the volumetric data. I tried Actions - Atoms/Bonds - Show, but I still don't see the atoms. Is there another step I should take to make them visible?
I see something like this:
<PastedGraphic-1.png>
(very nice!)
I'm attaching a link to the cube file below -- I can't include it because it is 600 MB.
Thanks,
Zack
https://www.dropbox.com/s/ehhli8h3qr7vd4y/wfc_k001_b329wfc.cube?dl=0 <https://www.dropbox.com/s/ehhli8h3qr7vd4y/wfc_k001_b329wfc.cube?dl=0>
_______________________________________________ ChimeraX-users mailing list ChimeraX-users@cgl.ucsf.edu <mailto:ChimeraX-users@cgl.ucsf.edu> Manage subscription: https://www.rbvi.ucsf.edu/mailman/listinfo/chimerax-users
{kind=link}
Hi Zack, There is a line of code in the Python script that makes the wrong bonds s.connect_structure() that you can comment out by putting a "#" character in front of it. # s.connect_structure() Then it will just create the atoms and not the bonds. Thanks for mentioning the wrong indentation -- there were tabs in the code in the web page that got translated into 4 spaces instead of 8 -- I have fixed it by using all spaces. I made a feature request to improve the Gaussian cube reader to also make the atoms. It is a bit tricky to squeeze in because the current code is a volume data reader that can only return volume data. Busy working on a grant now, so will have to wait. https://www.rbvi.ucsf.edu/trac/ChimeraX/ticket/4207 Tom
On Feb 2, 2021, at 11:39 AM, Zack Gainsforth <zackg@berkeley.edu> wrote:
Hi Tom,
Sorry if I got my cube files crossed up! I’m comparing several at the present so I must have just mixed it up.
The latest image you sent looks correct, except for the bonds as you note. (It’s a spinel.) However, the projected orbitals look correct, so the code is superimposing the volumetric data on the atomic data correctly. I tried it your script and it works terrifically! (Although I think some indentation got lost in the post on github.)
I simply turned off the metallic bonds and wound up with (yet another cube file):
<PastedGraphic-2.png>
Thank you much!
Cheers,
Zack
On Feb 2, 2021, at 11:19 AM, Tom Goddard <goddard@sonic.net <mailto:goddard@sonic.net>> wrote:
Hi Zack,
Elaine is right that ChimeraX only reads the map in Gaussian cube files, not the atoms. I was curious what your atoms looked like so I wrote a short Python script to make the atoms from your file and put it on the ChimeraX Recipes web site as an example of ChimeraX Python code.
https://rbvi.github.io/chimerax-recipes/gaussian_cube_atoms/gcatoms.html <https://rbvi.github.io/chimerax-recipes/gaussian_cube_atoms/gcatoms.html>
The Gaussian cube file you linked to in your original message is not the same as the one you show in your second message. In the linked file I see a lattice of aluminum, magnesium and oxygen. ChimeraX tries to make metal coordination bonds (purple dashed lines) and put bonds between various aluminum atoms and made a pretty bizarre and likely wrong picture. ChimeraX is designed for proteins so doesn't know what to do with inorganic crystals.
Tom
<wfc_K001_B329wfc.png>
On Feb 2, 2021, at 10:42 AM, Zack Gainsforth <zackg@berkeley.edu <mailto:zackg@berkeley.edu>> wrote:
Hi Elaine,
Thanks for the feedback!
I think it does contain atomic coordinates because if I open the file up, the first few lines say:
Cubefile created from PWScf calculation k_point 1, band 493 112 0.000000 0.000000 0.000000 360 0.000000 0.043010 0.043010 360 0.043010 0.000000 0.043010 360 0.043010 0.043010 0.000000 13 13.000000 15.550626 7.919837 23.249520 13 13.000000 7.794637 15.500714 23.173025 13 13.000000 15.344622 7.808598 7.696103 24 24.000000 8.153563 23.302893 15.676357 24 24.000000 22.842015 15.147909 23.267895 24 24.000000 23.276582 23.168925 15.892733 24 24.000000 0.413132 15.595221 15.687712 13 13.000000 15.337306 19.401078 19.211118 24 24.000000 23.238956 26.684104 19.354277 13 13.000000 30.836280 19.498740 19.483616 13 13.000000 23.156913 11.704068 19.344564 13 13.000000 7.845982 3.847472 11.610165
So we have 13 being magnesium, and 24 being Cr, etc.
After the 112 atoms, it starts with the volumetric values.
So it sounds like perhaps I should request a feature addition to ChimeraX to implement atomic coordinates as well?
Following your advice though, I saved the structure as a pdb also, and loaded that and it seems to work — though there is an offset between the volumetric and atomic datasets.
<PastedGraphic-1.png>
I guess I would need to manually shift one of them, so that provides a workaround for now.
Thanks,
Zack
On Feb 2, 2021, at 10:15 AM, Elaine Meng <meng@cgl.ucsf.edu <mailto:meng@cgl.ucsf.edu>> wrote:
Hi Zack, A cube file is a map format (values at points on a regular grid), and is not used for atomic coordinates. In other words, I believe your file contains only the wavefunctions. You would need to also open a file for the atomic coordinates (possible formats PDB, SDF, MOL, ...) which you may have input to Quantum Espresso, or may be able to write from the program. I've never used it, so I don't know what it writes.
ChimeraX knows these atomic formats: <https://rbvi.ucsf.edu/chimerax/docs/user/commands/open.html#atomic <https://rbvi.ucsf.edu/chimerax/docs/user/commands/open.html#atomic>> ... and these map formats: <https://rbvi.ucsf.edu/chimerax/docs/user/commands/open.html#volume <https://rbvi.ucsf.edu/chimerax/docs/user/commands/open.html#volume>>
I hope this helps, Elaine ----- Elaine C. Meng, Ph.D. UCSF Chimera(X) team Department of Pharmaceutical Chemistry University of California, San Francisco
On Feb 2, 2021, at 9:45 AM, Zack Gainsforth <zackg@berkeley.edu <mailto:zackg@berkeley.edu>> wrote:
Hi,
I produced some projected wavefunctions in a crystal that I'm working with using Quantum Espresso (DFT). I output these projections to a gaussian cube file using the pp.x program (built into Quantum Espresso). I'm able to read these into ChimeraX but I'm unable to see the atoms -- only the volumetric data. I tried Actions - Atoms/Bonds - Show, but I still don't see the atoms. Is there another step I should take to make them visible?
I see something like this:
<PastedGraphic-1.png>
(very nice!)
I'm attaching a link to the cube file below -- I can't include it because it is 600 MB.
Thanks,
Zack
https://www.dropbox.com/s/ehhli8h3qr7vd4y/wfc_k001_b329wfc.cube?dl=0 <https://www.dropbox.com/s/ehhli8h3qr7vd4y/wfc_k001_b329wfc.cube?dl=0>
_______________________________________________ ChimeraX-users mailing list ChimeraX-users@cgl.ucsf.edu <mailto:ChimeraX-users@cgl.ucsf.edu> Manage subscription: https://www.rbvi.ucsf.edu/mailman/listinfo/chimerax-users <https://www.rbvi.ucsf.edu/mailman/listinfo/chimerax-users>
_______________________________________________ ChimeraX-users mailing list ChimeraX-users@cgl.ucsf.edu Manage subscription: https://www.rbvi.ucsf.edu/mailman/listinfo/chimerax-users
participants (3)
-
 Elaine Meng
Elaine Meng -
 Tom Goddard
Tom Goddard -
 Zack Gainsforth
Zack Gainsforth