ChimeraX: mmcif connectivity lost in some chains; OK in Coot

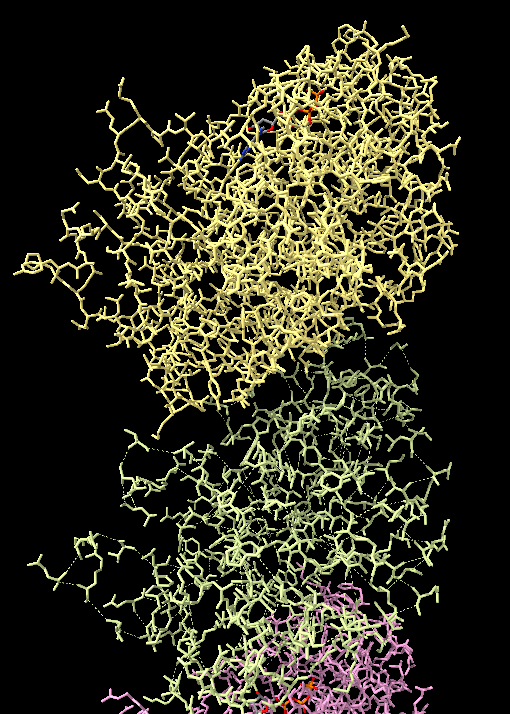
Dear ChimeraX team, Thank you very much for your work on this great software. We use it daily, and without which it would be extremely difficult to articulate our science. I am touching base with a problem I am having after migrating from a pdb --> mmcif format.For context, I am running ChimeraX on Ubuntu 20.04 using a fresh deb install this morning: chimerax --version UCSF ChimeraX version: 1.0 (2020-06-04) © 2016-2020 Regents of the University of California. All rights reserved. To be explicit, my problem is entirely reproducible. Specifically, I have a file (attached) that when opened in ccpem's Coot v0.9-pre, has all the connectivity present. However, when I switch to ChimeraX, the connectivity of some chains are lost. A good example of this contrast on chain connectivity fidelity is chain LA and LB. (photo attached). I tried reading through the ChimeraX documentation on this CIF format,<https://www.rbvi.ucsf.edu/chimerax/docs/devel/bundles/mmcif/src/mmcif_guidel...> but it remained not totally clear to me what is going awry in some chains but not in others. If you might have any insight on how to overcome this problem in chimerax, and allow all the chains to remain their connectivity, I would very much appreciate your thoughts/wisdom on the topic. ~Jacob [cid:0f6782e1-eb79-43db-93e8-0f8f44417fb3]
{kind=link}
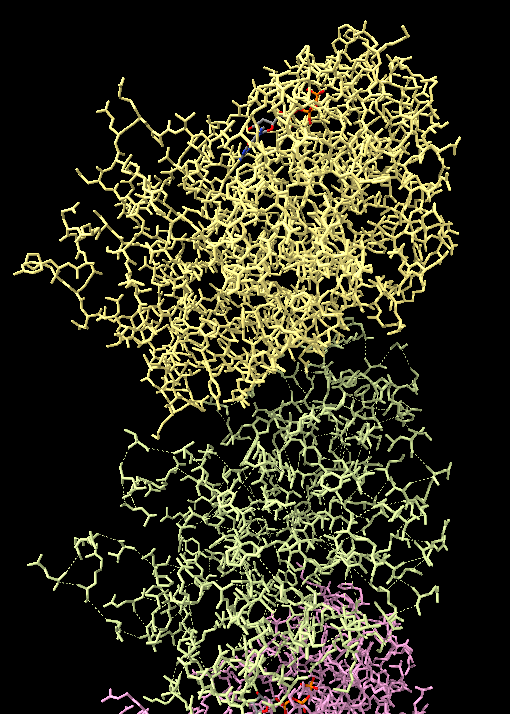
As an addendum, in coot, I saved OK mmcif file as a PDB (in zip as coot-mmcifTOpdb.pdb). Reviewing the file, it looks OK. It also opens and docks OK in ChimeraX with proper connectivity. However, upon saving the pdb with its updated coordinates after using the "Fit To Map" module, all the atoms have been changed to HETATM (in zip as it_to_map_PDB_savedinchimerax.pdb) Might this be a bug? or possibly a product of saving a cif file in coot to pdb? It was reproduced upon a second try. Maybe to consolidate the problems: 1) mmcif not connected in chimerax, but OK in coot [upon trying a workaround with an OK PDB file] 2) A fit to map PDB, when saved with chimerax, has all atoms changed to HETATM ~jacob ________________________________ From: Anderson, Jacob Sent: Saturday, June 13, 2020 1:48 PM To: chimerax-users@cgl.ucsf.edu <chimerax-users@cgl.ucsf.edu> Subject: ChimeraX: mmcif connectivity lost in some chains; OK in Coot Dear ChimeraX team, Thank you very much for your work on this great software. We use it daily, and without which it would be extremely difficult to articulate our science. I am touching base with a problem I am having after migrating from a pdb --> mmcif format.For context, I am running ChimeraX on Ubuntu 20.04 using a fresh deb install this morning: chimerax --version UCSF ChimeraX version: 1.0 (2020-06-04) © 2016-2020 Regents of the University of California. All rights reserved. To be explicit, my problem is entirely reproducible. Specifically, I have a file (attached) that when opened in ccpem's Coot v0.9-pre, has all the connectivity present. However, when I switch to ChimeraX, the connectivity of some chains are lost. A good example of this contrast on chain connectivity fidelity is chain LA and LB. (photo attached). I tried reading through the ChimeraX documentation on this CIF format,<https://www.rbvi.ucsf.edu/chimerax/docs/devel/bundles/mmcif/src/mmcif_guidel...> but it remained not totally clear to me what is going awry in some chains but not in others. If you might have any insight on how to overcome this problem in chimerax, and allow all the chains to remain their connectivity, I would very much appreciate your thoughts/wisdom on the topic. ~Jacob [cid:0f6782e1-eb79-43db-93e8-0f8f44417fb3]
{kind=link}

Hi Jacob, I can’t answer your first question (Greg Couch, our “mmCIF guy”, is the expert there), but I can help with the second. In the PDB standard there are 3 columns allocated to the residue name, followed by a blank column and then one column for the chain ID. Some programs steal that blank column for a 4-character residue name (Amber) and some steal it for a 2-character chain ID. ChimeraX’s PDB code is based on code from Chimera, and Chimera interfaced extensively with Amber programs and therefore interprets that column as as the 4th character of the residue name. So, when that file is read in you get single-character chain IDs (which is fine in your case since you only had ~10 chains that only differed in the second character) and residue names like SERL and ALAL. Obviously, those names are non-standard and therefore ChimeraX writes them out in HETATM records. The good news is that your problem is pretty easy to fix, at least in this particular case: just throw away the fourth character of the residue name. I have attached a simple Python script that does just that, which you just use the ‘open’ command to run (i.e. “open res-fix.py”). --Eric Eric Pettersen UCSF Computer Graphics Lab
On Jun 13, 2020, at 2:59 PM, Anderson, Jacob <jacob_r_anderson@hms.harvard.edu> wrote:
As an addendum, in coot, I saved OK mmcif file as a PDB (in zip as coot-mmcifTOpdb.pdb). Reviewing the file, it looks OK. It also opens and docks OK in ChimeraX with proper connectivity. However, upon saving the pdb with its updated coordinates after using the "Fit To Map" module, all the atoms have been changed to HETATM (in zip as it_to_map_PDB_savedinchimerax.pdb)
Might this be a bug? or possibly a product of saving a cif file in coot to pdb? It was reproduced upon a second try.
Maybe to consolidate the problems:
1) mmcif not connected in chimerax, but OK in coot [upon trying a workaround with an OK PDB file] 2) A fit to map PDB, when saved with chimerax, has all atoms changed to HETATM
~jacob From: Anderson, Jacob Sent: Saturday, June 13, 2020 1:48 PM To: chimerax-users@cgl.ucsf.edu <mailto:chimerax-users@cgl.ucsf.edu> <chimerax-users@cgl.ucsf.edu <mailto:chimerax-users@cgl.ucsf.edu>> Subject: ChimeraX: mmcif connectivity lost in some chains; OK in Coot
Dear ChimeraX team,
Thank you very much for your work on this great software. We use it daily, and without which it would be extremely difficult to articulate our science.
I am touching base with a problem I am having after migrating from a pdb --> mmcif format.For context, I am running ChimeraX on Ubuntu 20.04 using a fresh deb install this morning:
chimerax --version UCSF ChimeraX version: 1.0 (2020-06-04) © 2016-2020 Regents of the University of California. All rights reserved.
To be explicit, my problem is entirely reproducible. Specifically, I have a file (attached) that when opened in ccpem's Coot v0.9-pre, has all the connectivity present. However, when I switch to ChimeraX, the connectivity of some chains are lost. A good example of this contrast on chain connectivity fidelity is chain LA and LB. (photo attached).
I tried reading through the ChimeraX documentation on this CIF format, <https://www.rbvi.ucsf.edu/chimerax/docs/devel/bundles/mmcif/src/mmcif_guidel...> but it remained not totally clear to me what is going awry in some chains but not in others.
If you might have any insight on how to overcome this problem in chimerax, and allow all the chains to remain their connectivity, I would very much appreciate your thoughts/wisdom on the topic.
~Jacob
<image.png> <pdb_hetatm_files.zip>_______________________________________________ ChimeraX-users mailing list ChimeraX-users@cgl.ucsf.edu <mailto:ChimeraX-users@cgl.ucsf.edu> Manage subscription: https://www.rbvi.ucsf.edu/mailman/listinfo/chimerax-users <https://www.rbvi.ucsf.edu/mailman/listinfo/chimerax-users>
I meant to also say that we have a ticket open in our bug-tracking database to make the handling of that blank column more flexible, and I will add you to the cc list for that ticket so that you’ll know when it gets worked on. —Eric
On Jun 15, 2020, at 10:11 AM, Eric Pettersen <pett@cgl.ucsf.edu> wrote:
Hi Jacob, I can’t answer your first question (Greg Couch, our “mmCIF guy”, is the expert there), but I can help with the second. In the PDB standard there are 3 columns allocated to the residue name, followed by a blank column and then one column for the chain ID. Some programs steal that blank column for a 4-character residue name (Amber) and some steal it for a 2-character chain ID. ChimeraX’s PDB code is based on code from Chimera, and Chimera interfaced extensively with Amber programs and therefore interprets that column as as the 4th character of the residue name. So, when that file is read in you get single-character chain IDs (which is fine in your case since you only had ~10 chains that only differed in the second character) and residue names like SERL and ALAL. Obviously, those names are non-standard and therefore ChimeraX writes them out in HETATM records. The good news is that your problem is pretty easy to fix, at least in this particular case: just throw away the fourth character of the residue name. I have attached a simple Python script that does just that, which you just use the ‘open’ command to run (i.e. “open res-fix.py”).
--Eric
Eric Pettersen UCSF Computer Graphics Lab
<res-fix.py>
On Jun 13, 2020, at 2:59 PM, Anderson, Jacob <jacob_r_anderson@hms.harvard.edu <mailto:jacob_r_anderson@hms.harvard.edu>> wrote:
As an addendum, in coot, I saved OK mmcif file as a PDB (in zip as coot-mmcifTOpdb.pdb). Reviewing the file, it looks OK. It also opens and docks OK in ChimeraX with proper connectivity. However, upon saving the pdb with its updated coordinates after using the "Fit To Map" module, all the atoms have been changed to HETATM (in zip as it_to_map_PDB_savedinchimerax.pdb)
Might this be a bug? or possibly a product of saving a cif file in coot to pdb? It was reproduced upon a second try.
Maybe to consolidate the problems:
1) mmcif not connected in chimerax, but OK in coot [upon trying a workaround with an OK PDB file] 2) A fit to map PDB, when saved with chimerax, has all atoms changed to HETATM
~jacob From: Anderson, Jacob Sent: Saturday, June 13, 2020 1:48 PM To: chimerax-users@cgl.ucsf.edu <mailto:chimerax-users@cgl.ucsf.edu> <chimerax-users@cgl.ucsf.edu <mailto:chimerax-users@cgl.ucsf.edu>> Subject: ChimeraX: mmcif connectivity lost in some chains; OK in Coot
Dear ChimeraX team,
Thank you very much for your work on this great software. We use it daily, and without which it would be extremely difficult to articulate our science.
I am touching base with a problem I am having after migrating from a pdb --> mmcif format.For context, I am running ChimeraX on Ubuntu 20.04 using a fresh deb install this morning:
chimerax --version UCSF ChimeraX version: 1.0 (2020-06-04) © 2016-2020 Regents of the University of California. All rights reserved.
To be explicit, my problem is entirely reproducible. Specifically, I have a file (attached) that when opened in ccpem's Coot v0.9-pre, has all the connectivity present. However, when I switch to ChimeraX, the connectivity of some chains are lost. A good example of this contrast on chain connectivity fidelity is chain LA and LB. (photo attached).
I tried reading through the ChimeraX documentation on this CIF format, <https://www.rbvi.ucsf.edu/chimerax/docs/devel/bundles/mmcif/src/mmcif_guidel...> but it remained not totally clear to me what is going awry in some chains but not in others.
If you might have any insight on how to overcome this problem in chimerax, and allow all the chains to remain their connectivity, I would very much appreciate your thoughts/wisdom on the topic.
~Jacob
<image.png> <pdb_hetatm_files.zip>_______________________________________________ ChimeraX-users mailing list ChimeraX-users@cgl.ucsf.edu <mailto:ChimeraX-users@cgl.ucsf.edu> Manage subscription: https://www.rbvi.ucsf.edu/mailman/listinfo/chimerax-users <https://www.rbvi.ucsf.edu/mailman/listinfo/chimerax-users>
_______________________________________________ ChimeraX-users mailing list ChimeraX-users@cgl.ucsf.edu Manage subscription: https://www.rbvi.ucsf.edu/mailman/listinfo/chimerax-users
Dear Dr. Pettersen, Thank you so much for your help. That helps me understand what was causing the problem greatly! Unfortunately this is just one filament of a large microtubule complex which we will have >100 chains (the reason we tried to move to mmcif). I wrote something to make it ATOM-ee so I could refine it in coot/phenix. I will probably continue with that pipeline (dock in chimerax, change to ATOM w/ python, refine in coot/phenix, wash and repeat). Perhaps if I figure out the mmcif issue it will be more OK. Thank you again for adding me to that thread! ~jacob ________________________________ From: Eric Pettersen <pett@cgl.ucsf.edu> Sent: Monday, June 15, 2020 1:11 PM To: Anderson, Jacob <jacob_r_anderson@hms.harvard.edu> Cc: chimerax-users@cgl.ucsf.edu <chimerax-users@cgl.ucsf.edu> Subject: Re: [chimerax-users] ChimeraX: mmcif connectivity lost in some chains; OK in Coot Hi Jacob, I can’t answer your first question (Greg Couch, our “mmCIF guy”, is the expert there), but I can help with the second. In the PDB standard there are 3 columns allocated to the residue name, followed by a blank column and then one column for the chain ID. Some programs steal that blank column for a 4-character residue name (Amber) and some steal it for a 2-character chain ID. ChimeraX’s PDB code is based on code from Chimera, and Chimera interfaced extensively with Amber programs and therefore interprets that column as as the 4th character of the residue name. So, when that file is read in you get single-character chain IDs (which is fine in your case since you only had ~10 chains that only differed in the second character) and residue names like SERL and ALAL. Obviously, those names are non-standard and therefore ChimeraX writes them out in HETATM records. The good news is that your problem is pretty easy to fix, at least in this particular case: just throw away the fourth character of the residue name. I have attached a simple Python script that does just that, which you just use the ‘open’ command to run (i.e. “open res-fix.py”). --Eric Eric Pettersen UCSF Computer Graphics Lab On Jun 13, 2020, at 2:59 PM, Anderson, Jacob <jacob_r_anderson@hms.harvard.edu<mailto:jacob_r_anderson@hms.harvard.edu>> wrote: As an addendum, in coot, I saved OK mmcif file as a PDB (in zip as coot-mmcifTOpdb.pdb). Reviewing the file, it looks OK. It also opens and docks OK in ChimeraX with proper connectivity. However, upon saving the pdb with its updated coordinates after using the "Fit To Map" module, all the atoms have been changed to HETATM (in zip as it_to_map_PDB_savedinchimerax.pdb) Might this be a bug? or possibly a product of saving a cif file in coot to pdb? It was reproduced upon a second try. Maybe to consolidate the problems: 1) mmcif not connected in chimerax, but OK in coot [upon trying a workaround with an OK PDB file] 2) A fit to map PDB, when saved with chimerax, has all atoms changed to HETATM ~jacob ________________________________ From: Anderson, Jacob Sent: Saturday, June 13, 2020 1:48 PM To: chimerax-users@cgl.ucsf.edu<mailto:chimerax-users@cgl.ucsf.edu> <chimerax-users@cgl.ucsf.edu<mailto:chimerax-users@cgl.ucsf.edu>> Subject: ChimeraX: mmcif connectivity lost in some chains; OK in Coot Dear ChimeraX team, Thank you very much for your work on this great software. We use it daily, and without which it would be extremely difficult to articulate our science. I am touching base with a problem I am having after migrating from a pdb --> mmcif format.For context, I am running ChimeraX on Ubuntu 20.04 using a fresh deb install this morning: chimerax --version UCSF ChimeraX version: 1.0 (2020-06-04) © 2016-2020 Regents of the University of California. All rights reserved. To be explicit, my problem is entirely reproducible. Specifically, I have a file (attached) that when opened in ccpem's Coot v0.9-pre, has all the connectivity present. However, when I switch to ChimeraX, the connectivity of some chains are lost. A good example of this contrast on chain connectivity fidelity is chain LA and LB. (photo attached). I tried reading through the ChimeraX documentation on this CIF format,<https://urldefense.proofpoint.com/v2/url?u=https-3A__www.rbvi.ucsf.edu_chime...> but it remained not totally clear to me what is going awry in some chains but not in others. If you might have any insight on how to overcome this problem in chimerax, and allow all the chains to remain their connectivity, I would very much appreciate your thoughts/wisdom on the topic. ~Jacob <image.png> <pdb_hetatm_files.zip>_______________________________________________ ChimeraX-users mailing list ChimeraX-users@cgl.ucsf.edu<mailto:ChimeraX-users@cgl.ucsf.edu> Manage subscription: https://www.rbvi.ucsf.edu/mailman/listinfo/chimerax-users<https://urldefense.proofpoint.com/v2/url?u=https-3A__plato.cgl.ucsf.edu_mailman_listinfo_chimerax-2Dusers&d=DwMFaQ&c=WO-RGvefibhHBZq3fL85hQ&r=WAfw3qrUG1ShU2YpwE9tWl1TX9X2iVUkDLLxcqiRjEo&m=Vg4JgEdu2yfdqjQKWHvvz4CoIUTuDldwAMK6apyPSrA&s=TqPt2UK_I5TXkKmACbYqexgynVNPJ28_fsYTqyWza3w&e=>

There are many problems with the given mmCIF file. The major problem for ChimeraX is that in the atom_site table, all of the chains are designated as the same entity, but they have different sequences. It would be best if you fixed the program that generated the mmCIF file to give each chain its own entity. It would be even better if the entity, entity_poly, and entity_poly_seq tables were given. A secondary problem is that the struct_conn table is malformed. The id is not unique. LINK is not a legal conn_type_id. The mandatory ptrn[12]_label_* columns are missing. The ptrn[12]_auth_* columns are given, but ptrn[12]_auth_atom_id's have explicit spaces them that don't match the atom_site.auth_atom_id's. And the ptrn[1]_symmetry fields are bogus. Again, it would be best if you fixed the program that generated the mmCIF file. That said, while I am reluctant to put in fixes for bad data because it slows down the mmCIF reader for everyone, I will see what I can do for the atom_site table. The struct_conn table is too messed up to workaround. HTH, Greg On 6/15/2020 10:21 AM, Anderson, Jacob wrote:
Dear Dr. Pettersen,
Thank you so much for your help. That helps me understand what was causing the problem greatly! Unfortunately this is just one filament of a large microtubule complex which we will have >100 chains (the reason we tried to move to mmcif).
I wrote something to make it ATOM-ee so I could refine it in coot/phenix. I will probably continue with that pipeline (dock in chimerax, change to ATOM w/ python, refine in coot/phenix, wash and repeat). Perhaps if I figure out the mmcif issue it will be more OK.
Thank you again for adding me to that thread!
~jacob
------------------------------------------------------------------------ *From:* Eric Pettersen <pett@cgl.ucsf.edu> *Sent:* Monday, June 15, 2020 1:11 PM *To:* Anderson, Jacob <jacob_r_anderson@hms.harvard.edu> *Cc:* chimerax-users@cgl.ucsf.edu <chimerax-users@cgl.ucsf.edu> *Subject:* Re: [chimerax-users] ChimeraX: mmcif connectivity lost in some chains; OK in Coot Hi Jacob, I can’t answer your first question (Greg Couch, our “mmCIF guy”, is the expert there), but I can help with the second. In the PDB standard there are 3 columns allocated to the residue name, followed by a blank column and then one column for the chain ID. Some programs steal that blank column for a 4-character residue name (Amber) and some steal it for a 2-character chain ID. ChimeraX’s PDB code is based on code from Chimera, and Chimera interfaced extensively with Amber programs and therefore interprets that column as as the 4th character of the residue name. So, when that file is read in you get single-character chain IDs (which is fine in your case since you only had ~10 chains that only differed in the second character) and residue names like SERL and ALAL. Obviously, those names are non-standard and therefore ChimeraX writes them out in HETATM records. The good news is that your problem is pretty easy to fix, at least in this particular case: just throw away the fourth character of the residue name. I have attached a simple Python script that does just that, which you just use the ‘open’ command to run (/i.e./ “open res-fix.py”).
--Eric
Eric Pettersen UCSF Computer Graphics Lab
On Jun 13, 2020, at 2:59 PM, Anderson, Jacob <jacob_r_anderson@hms.harvard.edu <mailto:jacob_r_anderson@hms.harvard.edu>> wrote:
As an addendum, in coot, I saved OK mmcif file as a PDB (in zip as coot-mmcifTOpdb.pdb). Reviewing the file, it looks OK. It also opens and docks OK in ChimeraX with proper connectivity. However, upon saving the pdb with its updated coordinates after using the "Fit To Map" module, all the atoms have been changed to HETATM (in zip as it_to_map_PDB_savedinchimerax.pdb)
Might this be a bug? or possibly a product of saving a cif file in coot to pdb? It was reproduced upon a second try.
Maybe to consolidate the problems:
1) mmcif not connected in chimerax, but OK in coot [upon trying a workaround with an OK PDB file] 2) A fit to map PDB, when saved with chimerax, has all atoms changed to HETATM
*~jacob*
------------------------------------------------------------------------ *From:*Anderson, Jacob *Sent:*Saturday, June 13, 2020 1:48 PM *To:*chimerax-users@cgl.ucsf.edu <mailto:chimerax-users@cgl.ucsf.edu><chimerax-users@cgl.ucsf.edu <mailto:chimerax-users@cgl.ucsf.edu>> *Subject:*ChimeraX: mmcif connectivity lost in some chains; OK in Coot Dear ChimeraX team,
Thank you very much for your work on this great software. We use it daily, and without which it would be extremely difficult to articulate our science.
I am touching base with a problem I am having after migrating from a pdb --> mmcif format.For context, I am running ChimeraX on Ubuntu 20.04 using a fresh deb install this morning:
chimerax --version UCSF ChimeraX version: 1.0 (2020-06-04) © 2016-2020 Regents of the University of California. All rights reserved.
To be explicit, my problem is entirely reproducible. Specifically, I have a file (attached) that when opened in ccpem's Coot v0.9-pre, has all the connectivity present. However, when I switch to ChimeraX, the connectivity of some chains are lost. A good example of this contrast on chain connectivity fidelity is chain LA and LB. (photo attached).
I tried reading through theChimeraX documentation on this CIF format, <https://urldefense.proofpoint.com/v2/url?u=https-3A__www.rbvi.ucsf.edu_chime...> but it remained not totally clear to me what is going awry in some chains but not in others.
If you might have any insight on how to overcome this problem in chimerax, and allow all the chains to remain their connectivity, I would very much appreciate your thoughts/wisdom on the topic.
~Jacob
<image.png>
<pdb_hetatm_files.zip>_______________________________________________ ChimeraX-users mailing list ChimeraX-users@cgl.ucsf.edu <mailto:ChimeraX-users@cgl.ucsf.edu> Manage subscription: https://www.rbvi.ucsf.edu/mailman/listinfo/chimerax-users <https://urldefense.proofpoint.com/v2/url?u=https-3A__plato.cgl.ucsf.edu_mail...>
_______________________________________________ ChimeraX-users mailing list ChimeraX-users@cgl.ucsf.edu Manage subscription: https://www.rbvi.ucsf.edu/mailman/listinfo/chimerax-users
participants (3)
-
 Anderson, Jacob
Anderson, Jacob -
 Eric Pettersen
Eric Pettersen -
 Greg Couch
Greg Couch