
Hello, My name is Diego. I’m a beginner user of ChimeraX (1.8) and I was wondering if there is an option to check the % of similarity between two structures after making the matchmaker. I mean, I make a matchmaker with two structures, can I see the % of similarity between these two structures? Not between the sequences, but between the structures. Thank you very much for your time and attention. Best regards Dr. Diego Angosto Bazarra Molecular inflammation and experimental surgery group. IMIB. University Hospital Virgen de la Arrixaca

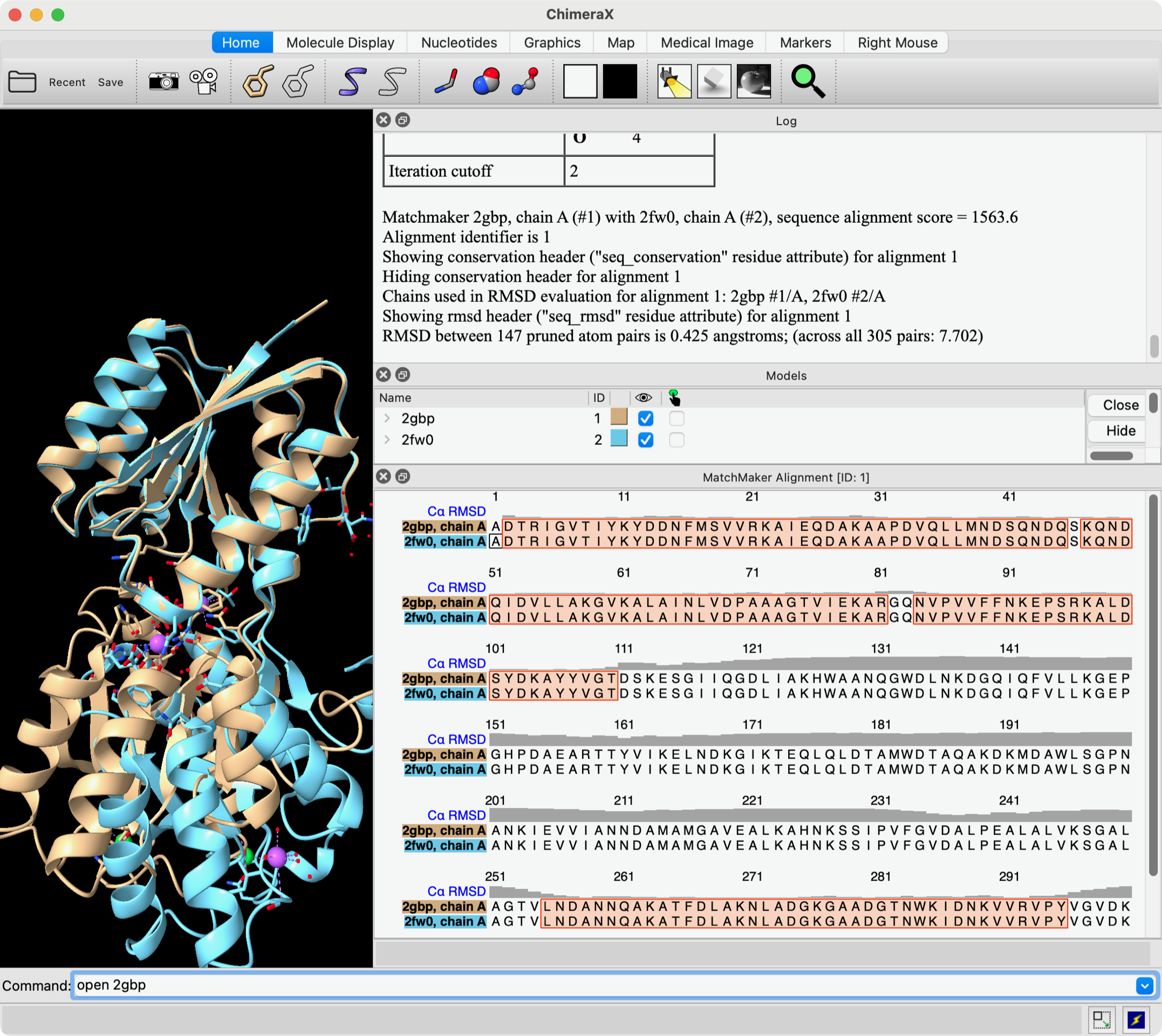
Hi Diego, As far as I know there is no measurement (in any program, I mean) that is the % similarity in structures. There are lots of other structure similarity measures. What people use the most is probably RMSD and that is already reported in the Log after you use matchmaker. Matchmaker uses only one atom per residue, CA atoms in amino acids, to calculate this RMSD. For example, these commands: open 2gbp open 2fw0 mm #2 to #1 show true ... open two structures and run Matchmaker, and result is shown in this screenshot. The "show true" shows the sequence alignment and the positions used for the final fit are outlined in orange boxes on the alignment. The Log window shows that the RMSD for the 147 positions (CA-CA pairs) in the orange boxes is 0.425 angstroms, and also across the entire sequences (305 CA-CA) pairs is 7.702 angstroms. The gray histogram over the sequences shows the CA-CA distance at each position. If you want to use more atoms (not just one CA atom per residue) to calculate the RMSD without moving the structures from their current positions, then you could use the "rmsd" command. See: <https://rbvi.ucsf.edu/chimerax/docs/user/commands/rmsd.html> Elaine ----- Elaine C. Meng, Ph.D. UCSF Chimera(X) team Resource for Biocomputing, Visualization, and Informatics Department of Pharmaceutical Chemistry University of California, San Francisco
On Jul 2, 2024, at 2:12 AM, DIEGO ANGOSTO BAZARRA via ChimeraX-users <chimerax-users@cgl.ucsf.edu> wrote:
Hello,
My name is Diego. I’m a beginner user of ChimeraX (1.8) and I was wondering if there is an option to check the % of similarity between two structures after making the matchmaker.
I mean, I make a matchmaker with two structures, can I see the % of similarity between these two structures? Not between the sequences, but between the structures.
Thank you very much for your time and attention.
Best regards
Dr. Diego Angosto Bazarra Molecular inflammation and experimental surgery group. IMIB. University Hospital Virgen de la Arrixaca
{kind=link}
Hi Elaine, Thank you very much for your answer, it is very helpful.!😁 I have another doubt: I use Chimera to predict structures using alphafold. When the session is completed, I get a folder with the different PDBs. Usually these PDBs are named as: af1024_unrelaxed_rank_001_alphafold2_ptm_model_4_seed_000.pdb , I get 5 different PDBs whose rank is between 001 and 005. Then I have a best model.pdb, Is this best model the rank_001.pdb? Would I be doing something wrong if I choose a different rank that matches more closely with lab data where I can calculate whether a structure is open or closed? I mean, I make the prediction, the best model gives me a "closed" structure and my lab data indicates an "open" structure. But a pdb with rank_002 or _003 shows open structure, is it possible to choose that model before than best model? I don't know if I have explained myself, my English is not very good 😅. Thank you very much for your time and help. Best Diego Dr. Diego Angosto Bazarra Molecular inflammation and experimental surgery group. IMIB. University Hospital Virgen de la Arrixaca El 2 jul 2024, a las 18:17, Elaine Meng <meng@cgl.ucsf.edu> escribió: Hi Diego, As far as I know there is no measurement (in any program, I mean) that is the % similarity in structures. There are lots of other structure similarity measures. What people use the most is probably RMSD and that is already reported in the Log after you use matchmaker. Matchmaker uses only one atom per residue, CA atoms in amino acids, to calculate this RMSD. For example, these commands: open 2gbp open 2fw0 mm #2 to #1 show true ... open two structures and run Matchmaker, and result is shown in this screenshot. The "show true" shows the sequence alignment and the positions used for the final fit are outlined in orange boxes on the alignment. The Log window shows that the RMSD for the 147 positions (CA-CA pairs) in the orange boxes is 0.425 angstroms, and also across the entire sequences (305 CA-CA) pairs is 7.702 angstroms. The gray histogram over the sequences shows the CA-CA distance at each position. <Screen Shot 2024-07-02 at 9.14.41 AM.png> If you want to use more atoms (not just one CA atom per residue) to calculate the RMSD without moving the structures from their current positions, then you could use the "rmsd" command. See: <https://rbvi.ucsf.edu/chimerax/docs/user/commands/rmsd.html<https://urldefense.com/v3/__https://rbvi.ucsf.edu/chimerax/docs/user/commands/rmsd.html__;!!D9dNQwwGXtA!QufkmShUR976cQ4hLnCncLjpogMWYSsNtO1fwUKgPF68Pc4zo1O5souPhLHoIPxFZqEamAjvbxXkz2Q$>> Elaine ----- Elaine C. Meng, Ph.D. UCSF Chimera(X) team Resource for Biocomputing, Visualization, and Informatics Department of Pharmaceutical Chemistry University of California, San Francisco On Jul 2, 2024, at 2:12 AM, DIEGO ANGOSTO BAZARRA via ChimeraX-users <chimerax-users@cgl.ucsf.edu<mailto:chimerax-users@cgl.ucsf.edu>> wrote: Hello, My name is Diego. I’m a beginner user of ChimeraX (1.8) and I was wondering if there is an option to check the % of similarity between two structures after making the matchmaker. I mean, I make a matchmaker with two structures, can I see the % of similarity between these two structures? Not between the sequences, but between the structures. Thank you very much for your time and attention. Best regards Dr. Diego Angosto Bazarra Molecular inflammation and experimental surgery group. IMIB. University Hospital Virgen de la Arrixaca

Hi Diego, It is reasonable to use any of the 5 AlphaFold predicted models. AlphaFold gives confidence scores to each prediction that you can read about here https://www.rbvi.ucsf.edu/chimerax/data/pae-apr2022/pae.html When AlphaFold gives different conformations for the 5 predictions it usually has low confidence in all of them. You shoud not put too much faith in low confidence AlphaFold predictions. Instead treat them as suggesting of a possible conformation that could help guide you to get more reliable experimental information. For instance if you see some interesting hydrogen bond in a predicted conformation you could try a mutation experiment that disrupts that putative hydrogen bond and run an assay to see if it changes protein function. Tom
On Jul 2, 2024, at 10:11 PM, DIEGO ANGOSTO BAZARRA via ChimeraX-users <chimerax-users@cgl.ucsf.edu> wrote:
Hi Elaine,
Thank you very much for your answer, it is very helpful.!😁 I have another doubt: I use Chimera to predict structures using alphafold. When the session is completed, I get a folder with the different PDBs. Usually these PDBs are named as: af1024_unrelaxed_rank_001_alphafold2_ptm_model_4_seed_000.pdb , I get 5 different PDBs whose rank is between 001 and 005. Then I have a best model.pdb, Is this best model the rank_001.pdb? Would I be doing something wrong if I choose a different rank that matches more closely with lab data where I can calculate whether a structure is open or closed? I mean, I make the prediction, the best model gives me a "closed" structure and my lab data indicates an "open" structure. But a pdb with rank_002 or _003 shows open structure, is it possible to choose that model before than best model?
I don't know if I have explained myself, my English is not very good 😅. Thank you very much for your time and help.
Best
Diego
Dr. Diego Angosto Bazarra Molecular inflammation and experimental surgery group. IMIB. University Hospital Virgen de la Arrixaca
El 2 jul 2024, a las 18:17, Elaine Meng <meng@cgl.ucsf.edu> escribió:
Hi Diego, As far as I know there is no measurement (in any program, I mean) that is the % similarity in structures. There are lots of other structure similarity measures. What people use the most is probably RMSD and that is already reported in the Log after you use matchmaker. Matchmaker uses only one atom per residue, CA atoms in amino acids, to calculate this RMSD.
For example, these commands:
open 2gbp open 2fw0 mm #2 to #1 show true
... open two structures and run Matchmaker, and result is shown in this screenshot. The "show true" shows the sequence alignment and the positions used for the final fit are outlined in orange boxes on the alignment. The Log window shows that the RMSD for the 147 positions (CA-CA pairs) in the orange boxes is 0.425 angstroms, and also across the entire sequences (305 CA-CA) pairs is 7.702 angstroms. The gray histogram over the sequences shows the CA-CA distance at each position.
<Screen Shot 2024-07-02 at 9.14.41 AM.png>
If you want to use more atoms (not just one CA atom per residue) to calculate the RMSD without moving the structures from their current positions, then you could use the "rmsd" command. See: <https://rbvi.ucsf.edu/chimerax/docs/user/commands/rmsd.html <https://urldefense.com/v3/__https://rbvi.ucsf.edu/chimerax/docs/user/commands/rmsd.html__;!!D9dNQwwGXtA!QufkmShUR976cQ4hLnCncLjpogMWYSsNtO1fwUKgPF68Pc4zo1O5souPhLHoIPxFZqEamAjvbxXkz2Q$>>
Elaine ----- Elaine C. Meng, Ph.D. UCSF Chimera(X) team Resource for Biocomputing, Visualization, and Informatics Department of Pharmaceutical Chemistry University of California, San Francisco
On Jul 2, 2024, at 2:12 AM, DIEGO ANGOSTO BAZARRA via ChimeraX-users <chimerax-users@cgl.ucsf.edu <mailto:chimerax-users@cgl.ucsf.edu>> wrote:
Hello,
My name is Diego. I’m a beginner user of ChimeraX (1.8) and I was wondering if there is an option to check the % of similarity between two structures after making the matchmaker.
I mean, I make a matchmaker with two structures, can I see the % of similarity between these two structures? Not between the sequences, but between the structures.
Thank you very much for your time and attention.
Best regards
Dr. Diego Angosto Bazarra Molecular inflammation and experimental surgery group. IMIB. University Hospital Virgen de la Arrixaca
_______________________________________________ ChimeraX-users mailing list -- chimerax-users@cgl.ucsf.edu To unsubscribe send an email to chimerax-users-leave@cgl.ucsf.edu Archives: https://mail.cgl.ucsf.edu/mailman/archives/list/chimerax-users@cgl.ucsf.edu/
participants (3)
-
 DIEGO ANGOSTO BAZARRA
DIEGO ANGOSTO BAZARRA -
 Elaine Meng
Elaine Meng -
 Tom Goddard
Tom Goddard