Identifying symmetry of a model

Hi I am working with pdb id: 3cx5. This model is roughly C2 symmetric. Is there a way to identify the symmetry (or rough symmetry of a not-so symmetric model) of a pdb/ mmcif model and orient the symmetry axis to the axis perpendicular to the screen? On a similar note, say we select a 4 somewhat symmetry-related residues in this model and make 2 intersecting lines, is there a way to align these lines perpendicular to the perpendicular screen axis? Thanks Yaikhomba

Hi Yaikhomba, ChimeraX doesn't have anything to automatically identify the symmetry of an atomic structure. There is only "measure symmetry" for maps, which is also somewhat limited in assuming standard orientations, etc. <https://rbvi.ucsf.edu/chimerax/docs/user/commands/measure.html#symmetry> However, say you have identified the approximate symmetry by eye. One possibility for C2 symetry is to open two copies of the structure, and then superimpose so that one half in one copy is on top of the other half of the other copy, then measure the rotation between the two. However, if the symmetry is only approximate, it is your own judgment call of which atoms are best to match to which atoms to obtain the best superposition of the halves. Example: open 3cx5 open 3cx5 mm #1/D to #2/O measure rotation #1 to #2 ... the measurement shows an axis which is really two markers (fake atoms) connected by a link (bond). You can manually orient the view to put that axis along the line of sight. You may need to temporarily hide the atomic models (e.g. with checkboxes in the Models panel) to see it clearly. Then you can hide the axis model if you want and turn the atomic models display back on. Chimera had an "align" command to put two atoms or markers along the line of sight, but ChimeraX does not have that feature. (ChimeraX "align" does something different, it superimposes atoms. You could use it to do the superposition, or use "mm" aka "matchmaker" as in my example above) <https://rbvi.ucsf.edu/chimerax/docs/user/commands/matchmaker.html> <https://rbvi.ucsf.edu/chimerax/docs/user/commands/measure.html#rotation> For your 4 residues case, you could define a plane from those residues (displayed as a flat disc) with "define" and then (again manually) rotate the view so that the plane is perpendicular to the line of sight. You can hide atomic models and/or hide the plane at any step with the Model panel checkboxes, as above. <https://rbvi.ucsf.edu/chimerax/docs/user/commands/define.html#plane> I hope this helps, Elaine ----- Elaine C. Meng, Ph.D. UCSF Chimera(X) team Department of Pharmaceutical Chemistry University of California, San Francisco
On Jan 18, 2021, at 9:02 AM, Y. Mutum <ym337@cam.ac.uk> wrote:
Hi
I am working with pdb id: 3cx5. This model is roughly C2 symmetric. Is there a way to identify the symmetry (or rough symmetry of a not-so symmetric model) of a pdb/ mmcif model and orient the symmetry axis to the axis perpendicular to the screen?
On a similar note, say we select a 4 somewhat symmetry-related residues in this model and make 2 intersecting lines, is there a way to align these lines perpendicular to the perpendicular screen axis?
Thanks Yaikhomba
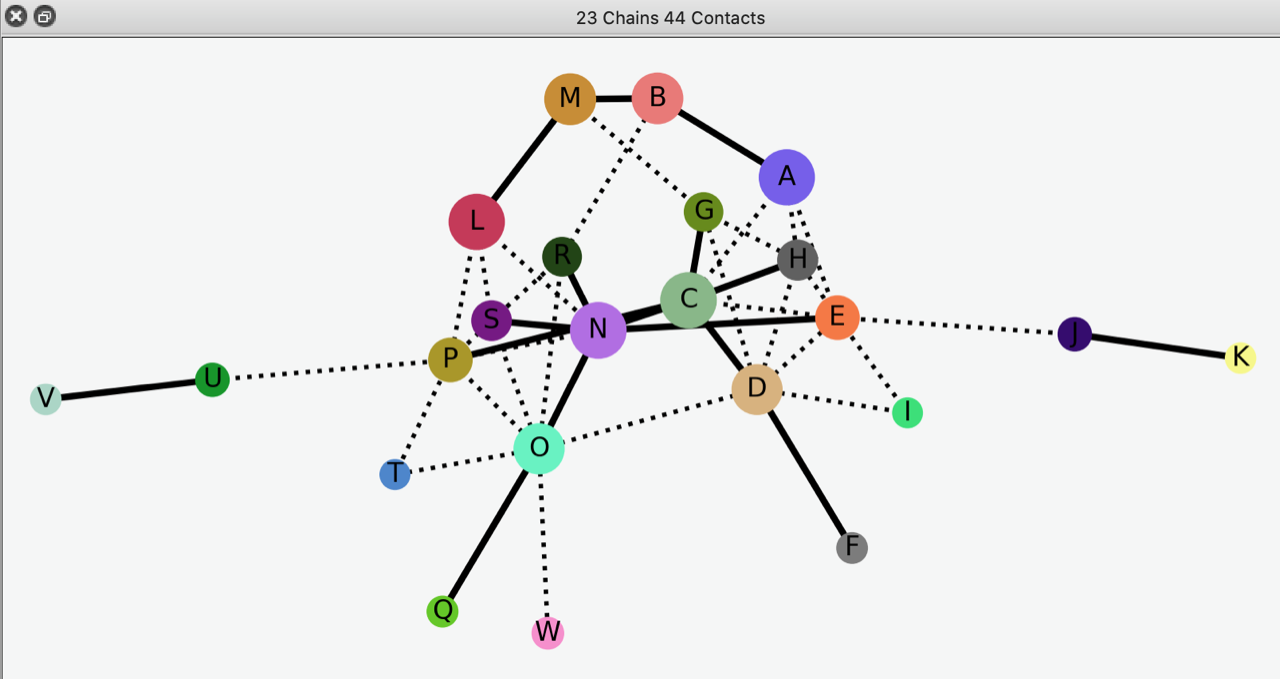
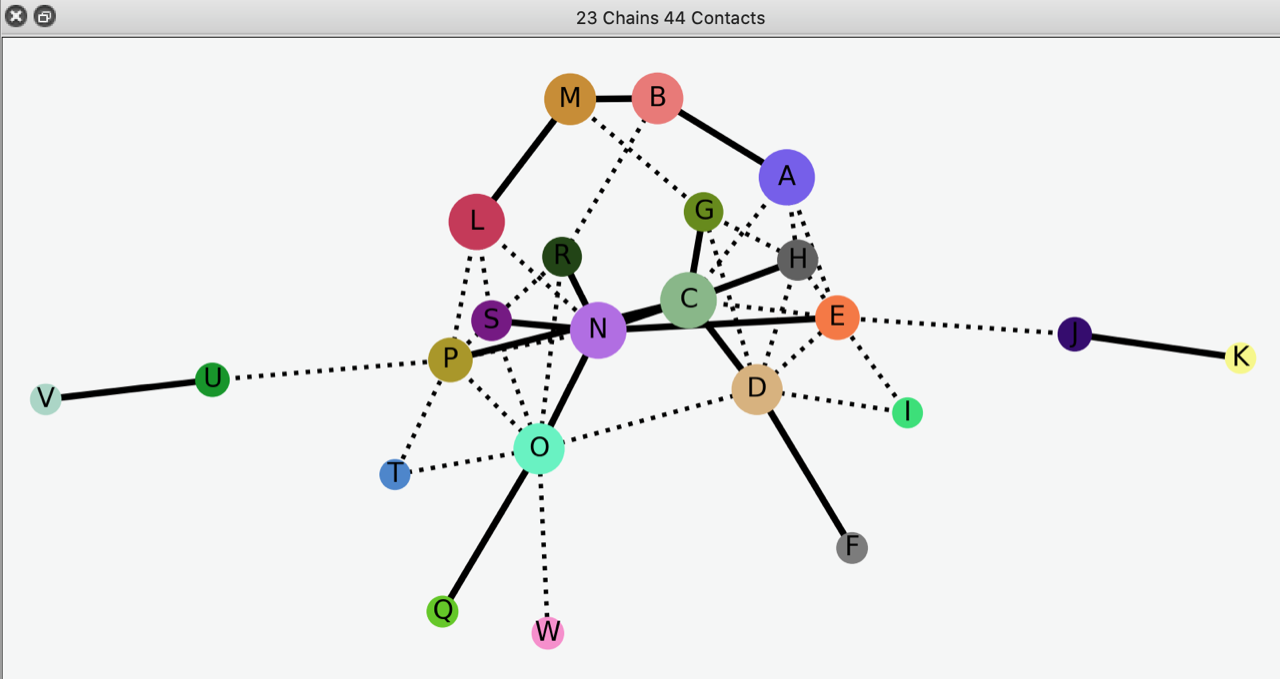
You probably already knew this, but the RCSB PDB reports symmetry and pseudosymmetry. E.g. 4hhb (alpha2 beta2 hemoglobin) is C2 symmetric and D2 pseudosymmetric. I also like the quaternary-structure diagrams shown in the 3D Complex database: <http://www.3dcomplex.org/> ChimeraX also shows subunit diagrams, and sometimes these help in understanding symmetry and/or pseudosymmetry of large complexes. For example, open 3cx5 interfaces protein <https://rbvi.ucsf.edu/chimerax/docs/user/commands/interfaces.html> ... it may take a while because it's based on calculated buried surface areas. A screenshot of the resulting interactive diagram is attached. Best, Elaine
On Jan 18, 2021, at 10:09 AM, Elaine Meng <meng@cgl.ucsf.edu> wrote:
Hi Yaikhomba, ChimeraX doesn't have anything to automatically identify the symmetry of an atomic structure. There is only "measure symmetry" for maps, which is also somewhat limited in assuming standard orientations, etc. <https://rbvi.ucsf.edu/chimerax/docs/user/commands/measure.html#symmetry>
However, say you have identified the approximate symmetry by eye. One possibility for C2 symetry is to open two copies of the structure, and then superimpose so that one half in one copy is on top of the other half of the other copy, then measure the rotation between the two. However, if the symmetry is only approximate, it is your own judgment call of which atoms are best to match to which atoms to obtain the best superposition of the halves.
Example: open 3cx5 open 3cx5 mm #1/D to #2/O measure rotation #1 to #2
... the measurement shows an axis which is really two markers (fake atoms) connected by a link (bond). You can manually orient the view to put that axis along the line of sight. You may need to temporarily hide the atomic models (e.g. with checkboxes in the Models panel) to see it clearly. Then you can hide the axis model if you want and turn the atomic models display back on.
Chimera had an "align" command to put two atoms or markers along the line of sight, but ChimeraX does not have that feature. (ChimeraX "align" does something different, it superimposes atoms. You could use it to do the superposition, or use "mm" aka "matchmaker" as in my example above)
<https://rbvi.ucsf.edu/chimerax/docs/user/commands/matchmaker.html> <https://rbvi.ucsf.edu/chimerax/docs/user/commands/measure.html#rotation>
For your 4 residues case, you could define a plane from those residues (displayed as a flat disc) with "define" and then (again manually) rotate the view so that the plane is perpendicular to the line of sight. You can hide atomic models and/or hide the plane at any step with the Model panel checkboxes, as above.
<https://rbvi.ucsf.edu/chimerax/docs/user/commands/define.html#plane>
I hope this helps, Elaine ----- Elaine C. Meng, Ph.D. UCSF Chimera(X) team Department of Pharmaceutical Chemistry University of California, San Francisco
On Jan 18, 2021, at 9:02 AM, Y. Mutum <ym337@cam.ac.uk> wrote:
Hi
I am working with pdb id: 3cx5. This model is roughly C2 symmetric. Is there a way to identify the symmetry (or rough symmetry of a not-so symmetric model) of a pdb/ mmcif model and orient the symmetry axis to the axis perpendicular to the screen?
On a similar note, say we select a 4 somewhat symmetry-related residues in this model and make 2 intersecting lines, is there a way to align these lines perpendicular to the perpendicular screen axis?
Thanks Yaikhomba
_______________________________________________ ChimeraX-users mailing list ChimeraX-users@cgl.ucsf.edu Manage subscription: https://www.rbvi.ucsf.edu/mailman/listinfo/chimerax-users
{kind=link}
Hi Elaine Thank you - both of them are indeed very helpful. I was not aware of the 4° structure diagrams - this might help. Thanks Yaikhomba Get Outlook for Android<https://aka.ms/ghei36> ________________________________ From: Elaine Meng <meng@cgl.ucsf.edu> Sent: Monday, January 18, 2021 6:28:15 PM To: Y. Mutum <ym337@cam.ac.uk> Cc: ChimeraX Users Help <chimerax-users@cgl.ucsf.edu> Subject: Re: [chimerax-users] Identifying symmetry of a model You probably already knew this, but the RCSB PDB reports symmetry and pseudosymmetry. E.g. 4hhb (alpha2 beta2 hemoglobin) is C2 symmetric and D2 pseudosymmetric. I also like the quaternary-structure diagrams shown in the 3D Complex database: <http://www.3dcomplex.org/> ChimeraX also shows subunit diagrams, and sometimes these help in understanding symmetry and/or pseudosymmetry of large complexes. For example, open 3cx5 interfaces protein <https://rbvi.ucsf.edu/chimerax/docs/user/commands/interfaces.html> ... it may take a while because it's based on calculated buried surface areas. A screenshot of the resulting interactive diagram is attached. Best, Elaine [cid:55488F1B-3339-401B-A69D-AAC390C73A44@gateway.sonic.net] On Jan 18, 2021, at 10:09 AM, Elaine Meng <meng@cgl.ucsf.edu<mailto:meng@cgl.ucsf.edu>> wrote: Hi Yaikhomba, ChimeraX doesn't have anything to automatically identify the symmetry of an atomic structure. There is only "measure symmetry" for maps, which is also somewhat limited in assuming standard orientations, etc. <https://rbvi.ucsf.edu/chimerax/docs/user/commands/measure.html#symmetry> However, say you have identified the approximate symmetry by eye. One possibility for C2 symetry is to open two copies of the structure, and then superimpose so that one half in one copy is on top of the other half of the other copy, then measure the rotation between the two. However, if the symmetry is only approximate, it is your own judgment call of which atoms are best to match to which atoms to obtain the best superposition of the halves. Example: open 3cx5 open 3cx5 mm #1/D to #2/O measure rotation #1 to #2 ... the measurement shows an axis which is really two markers (fake atoms) connected by a link (bond). You can manually orient the view to put that axis along the line of sight. You may need to temporarily hide the atomic models (e.g. with checkboxes in the Models panel) to see it clearly. Then you can hide the axis model if you want and turn the atomic models display back on. Chimera had an "align" command to put two atoms or markers along the line of sight, but ChimeraX does not have that feature. (ChimeraX "align" does something different, it superimposes atoms. You could use it to do the superposition, or use "mm" aka "matchmaker" as in my example above) <https://rbvi.ucsf.edu/chimerax/docs/user/commands/matchmaker.html> <https://rbvi.ucsf.edu/chimerax/docs/user/commands/measure.html#rotation> For your 4 residues case, you could define a plane from those residues (displayed as a flat disc) with "define" and then (again manually) rotate the view so that the plane is perpendicular to the line of sight. You can hide atomic models and/or hide the plane at any step with the Model panel checkboxes, as above. <https://rbvi.ucsf.edu/chimerax/docs/user/commands/define.html#plane> I hope this helps, Elaine ----- Elaine C. Meng, Ph.D. UCSF Chimera(X) team Department of Pharmaceutical Chemistry University of California, San Francisco On Jan 18, 2021, at 9:02 AM, Y. Mutum <ym337@cam.ac.uk<mailto:ym337@cam.ac.uk>> wrote: Hi I am working with pdb id: 3cx5. This model is roughly C2 symmetric. Is there a way to identify the symmetry (or rough symmetry of a not-so symmetric model) of a pdb/ mmcif model and orient the symmetry axis to the axis perpendicular to the screen? On a similar note, say we select a 4 somewhat symmetry-related residues in this model and make 2 intersecting lines, is there a way to align these lines perpendicular to the perpendicular screen axis? Thanks Yaikhomba _______________________________________________ ChimeraX-users mailing list ChimeraX-users@cgl.ucsf.edu<mailto:ChimeraX-users@cgl.ucsf.edu> Manage subscription: https://www.rbvi.ucsf.edu/mailman/listinfo/chimerax-users
{kind=link}
Hi Elaine I was thinking of a follow-up on this question, as this question is linked to the previous one. This answer might be slightly subjective, but given your ChimeraX expertise in visual representation, your thoughts would nonetheless be helpful. So, now that we have aligned the rotation axis perpendicular to the screen (from the previous question), I am thinking of putting in a small scalebar - say, 50 A. I created this rectange object (command: shape rectange width 50 height 2), along with the rotation axis and the other 3cx5.pdb's in ChimeraX(the email below) 1. Where would you recommend placing the scale bar (in this case, a rectange) wrt the model 3cx5, considering we want a view perpendicular the rotation axis, (showing the whole protein-complex to get a genral ide of the size of the object)? (considering that a distance is defined for in a plane). 2. The other related question is - if suppose, we draw a line b/w the Fe-centres of the hemes (by distance command from #1/C:4002 and #2/C:4002), how do we align the rectange object against this distance line. I can think of doing aligning it manually, but this might not be entirely accurate and I am not sure if there's some other method - such as aligning this rectange perpendicular to the rotation-axis line (generated from the previous email). I am also open to consider other alternatives, rather than this method as well. Thanks Yaikhomba ________________________________ From: Elaine Meng <meng@cgl.ucsf.edu> Sent: 18 January 2021 18:09 To: Y. Mutum <ym337@cam.ac.uk> Cc: chimerax-users@cgl.ucsf.edu <chimerax-users@cgl.ucsf.edu> Subject: Re: [chimerax-users] Identifying symmetry of a model Hi Yaikhomba, ChimeraX doesn't have anything to automatically identify the symmetry of an atomic structure. There is only "measure symmetry" for maps, which is also somewhat limited in assuming standard orientations, etc. <https://rbvi.ucsf.edu/chimerax/docs/user/commands/measure.html#symmetry> However, say you have identified the approximate symmetry by eye. One possibility for C2 symetry is to open two copies of the structure, and then superimpose so that one half in one copy is on top of the other half of the other copy, then measure the rotation between the two. However, if the symmetry is only approximate, it is your own judgment call of which atoms are best to match to which atoms to obtain the best superposition of the halves. Example: open 3cx5 open 3cx5 mm #1/D to #2/O measure rotation #1 to #2 ... the measurement shows an axis which is really two markers (fake atoms) connected by a link (bond). You can manually orient the view to put that axis along the line of sight. You may need to temporarily hide the atomic models (e.g. with checkboxes in the Models panel) to see it clearly. Then you can hide the axis model if you want and turn the atomic models display back on. Chimera had an "align" command to put two atoms or markers along the line of sight, but ChimeraX does not have that feature. (ChimeraX "align" does something different, it superimposes atoms. You could use it to do the superposition, or use "mm" aka "matchmaker" as in my example above) <https://rbvi.ucsf.edu/chimerax/docs/user/commands/matchmaker.html> <https://rbvi.ucsf.edu/chimerax/docs/user/commands/measure.html#rotation> For your 4 residues case, you could define a plane from those residues (displayed as a flat disc) with "define" and then (again manually) rotate the view so that the plane is perpendicular to the line of sight. You can hide atomic models and/or hide the plane at any step with the Model panel checkboxes, as above. <https://rbvi.ucsf.edu/chimerax/docs/user/commands/define.html#plane> I hope this helps, Elaine ----- Elaine C. Meng, Ph.D. UCSF Chimera(X) team Department of Pharmaceutical Chemistry University of California, San Francisco
On Jan 18, 2021, at 9:02 AM, Y. Mutum <ym337@cam.ac.uk> wrote:
Hi
I am working with pdb id: 3cx5. This model is roughly C2 symmetric. Is there a way to identify the symmetry (or rough symmetry of a not-so symmetric model) of a pdb/ mmcif model and orient the symmetry axis to the axis perpendicular to the screen?
On a similar note, say we select a 4 somewhat symmetry-related residues in this model and make 2 intersecting lines, is there a way to align these lines perpendicular to the perpendicular screen axis?
Thanks Yaikhomba
Hi Yaikhomba, Unfortunately "shape rectangle" is a very difficult way to try to make a scale bar because of these alignment issues: it does not have an option to align according to certain atoms or to the screen coordinate system. I can only try to rotate and translate it independently using the "move selected models" mode after creating it, and it is extremely difficult to get the rotation right by hand and eye. So you may need to create a scale bar using some other image-editing program after saving the image. ChimeraX command "zoom" without any other words reports the pixel size at center of rotation, same as overall if you are using orthographic projection (see #1 below). I.e. that is the conversion factor between window/image size and physical distance. However, I will try to answer the specific questions here: (1) I do not have a definitive answer for this, other than to say that when you are using perspective projection (the default), the nearer parts of the view are enlarged relative to the farther away parts. So I think it would be difficult to figure out exactly the correct "depth" to put this scale bar rectangle. Instead you may want to use orthographic projection which will not show nearer parts as larger, so that the scale bar would match all the parts, not just a certain depth. You can change from perspective to orthographic with command: camera ortho <https://rbvi.ucsf.edu/chimerax/docs/user/commands/camera.html> Now you will see just rectangles instead of the diverging lines in the Side View: tool show "Side View" (if you later want to change back to perspective, command: camera mono ) (2) You could first use the new "zalign" option of "view" (**for this you would need a recent daily build**) to align the two iron atoms along the screen Z-axis (in/out of screen), and then use "turn y 90" to rotate 90 degrees around screen Y axis (vertical). E.g. select the two iron atoms, and then view zalign sel turn y 90 You might want to use a different specification after the "view" part, e.g. similar to these commands: view #1 zalign sel view protein zalign sel view sel :<8 zalign sel <https://rbvi.ucsf.edu/chimerax/docs/user/commands/view.html> I hope this helps, Elaine ----- Elaine C. Meng, Ph.D. UCSF Chimera(X) team Department of Pharmaceutical Chemistry University of California, San Francisco
On Feb 5, 2021, at 7:31 AM, Y. Mutum <ym337@cam.ac.uk> wrote:
Hi Elaine
I was thinking of a follow-up on this question, as this question is linked to the previous one.
This answer might be slightly subjective, but given your ChimeraX expertise in visual representation, your thoughts would nonetheless be helpful.
So, now that we have aligned the rotation axis perpendicular to the screen (from the previous question), I am thinking of putting in a small scalebar - say, 50 A. I created this rectange object (command: shape rectange width 50 height 2), along with the rotation axis and the other 3cx5.pdb's in ChimeraX(the email below) • Where would you recommend placing the scale bar (in this case, a rectange) wrt the model 3cx5, considering we want a view perpendicular the rotation axis, (showing the whole protein-complex to get a genral ide of the size of the object)? (considering that a distance is defined for in a plane). • The other related question is - if suppose, we draw a line b/w the Fe-centres of the hemes (by distance command from #1/C:4002 and #2/C:4002), how do we align the rectange object against this distance line. I can think of doing aligning it manually, but this might not be entirely accurate and I am not sure if there's some other method - such as aligning this rectange perpendicular to the rotation-axis line (generated from the previous email).
I am also open to consider other alternatives, rather than this method as well.
Thanks Yaikhomba From: Elaine Meng <meng@cgl.ucsf.edu> Sent: 18 January 2021 18:09 To: Y. Mutum <ym337@cam.ac.uk> Cc: chimerax-users@cgl.ucsf.edu <chimerax-users@cgl.ucsf.edu> Subject: Re: [chimerax-users] Identifying symmetry of a model
Hi Yaikhomba, ChimeraX doesn't have anything to automatically identify the symmetry of an atomic structure. There is only "measure symmetry" for maps, which is also somewhat limited in assuming standard orientations, etc. <https://rbvi.ucsf.edu/chimerax/docs/user/commands/measure.html#symmetry>
However, say you have identified the approximate symmetry by eye. One possibility for C2 symetry is to open two copies of the structure, and then superimpose so that one half in one copy is on top of the other half of the other copy, then measure the rotation between the two. However, if the symmetry is only approximate, it is your own judgment call of which atoms are best to match to which atoms to obtain the best superposition of the halves.
Example: open 3cx5 open 3cx5 mm #1/D to #2/O measure rotation #1 to #2
... the measurement shows an axis which is really two markers (fake atoms) connected by a link (bond). You can manually orient the view to put that axis along the line of sight. You may need to temporarily hide the atomic models (e.g. with checkboxes in the Models panel) to see it clearly. Then you can hide the axis model if you want and turn the atomic models display back on.
Chimera had an "align" command to put two atoms or markers along the line of sight, but ChimeraX does not have that feature. (ChimeraX "align" does something different, it superimposes atoms. You could use it to do the superposition, or use "mm" aka "matchmaker" as in my example above)
<https://rbvi.ucsf.edu/chimerax/docs/user/commands/matchmaker.html> <https://rbvi.ucsf.edu/chimerax/docs/user/commands/measure.html#rotation>
For your 4 residues case, you could define a plane from those residues (displayed as a flat disc) with "define" and then (again manually) rotate the view so that the plane is perpendicular to the line of sight. You can hide atomic models and/or hide the plane at any step with the Model panel checkboxes, as above.
<https://rbvi.ucsf.edu/chimerax/docs/user/commands/define.html#plane>
I hope this helps, Elaine ----- Elaine C. Meng, Ph.D. UCSF Chimera(X) team Department of Pharmaceutical Chemistry University of California, San Francisco
On Jan 18, 2021, at 9:02 AM, Y. Mutum <ym337@cam.ac.uk> wrote:
Hi
I am working with pdb id: 3cx5. This model is roughly C2 symmetric. Is there a way to identify the symmetry (or rough symmetry of a not-so symmetric model) of a pdb/ mmcif model and orient the symmetry axis to the axis perpendicular to the screen?
On a similar note, say we select a 4 somewhat symmetry-related residues in this model and make 2 intersecting lines, is there a way to align these lines perpendicular to the perpendicular screen axis?
Thanks Yaikhomba
_______________________________________________ ChimeraX-users mailing list ChimeraX-users@cgl.ucsf.edu Manage subscription: https://www.rbvi.ucsf.edu/mailman/listinfo/chimerax-users
Hi Elaine Thanks - this is a good workaround. Yaikhomba ________________________________ From: Elaine Meng <meng@cgl.ucsf.edu> Sent: 05 February 2021 17:55 To: Y. Mutum <ym337@cam.ac.uk> Cc: ChimeraX Users Help <chimerax-users@cgl.ucsf.edu> Subject: Re: [chimerax-users] Identifying symmetry of a model Hi Yaikhomba, Unfortunately "shape rectangle" is a very difficult way to try to make a scale bar because of these alignment issues: it does not have an option to align according to certain atoms or to the screen coordinate system. I can only try to rotate and translate it independently using the "move selected models" mode after creating it, and it is extremely difficult to get the rotation right by hand and eye. So you may need to create a scale bar using some other image-editing program after saving the image. ChimeraX command "zoom" without any other words reports the pixel size at center of rotation, same as overall if you are using orthographic projection (see #1 below). I.e. that is the conversion factor between window/image size and physical distance. However, I will try to answer the specific questions here: (1) I do not have a definitive answer for this, other than to say that when you are using perspective projection (the default), the nearer parts of the view are enlarged relative to the farther away parts. So I think it would be difficult to figure out exactly the correct "depth" to put this scale bar rectangle. Instead you may want to use orthographic projection which will not show nearer parts as larger, so that the scale bar would match all the parts, not just a certain depth. You can change from perspective to orthographic with command: camera ortho <https://rbvi.ucsf.edu/chimerax/docs/user/commands/camera.html> Now you will see just rectangles instead of the diverging lines in the Side View: tool show "Side View" (if you later want to change back to perspective, command: camera mono ) (2) You could first use the new "zalign" option of "view" (**for this you would need a recent daily build**) to align the two iron atoms along the screen Z-axis (in/out of screen), and then use "turn y 90" to rotate 90 degrees around screen Y axis (vertical). E.g. select the two iron atoms, and then view zalign sel turn y 90 You might want to use a different specification after the "view" part, e.g. similar to these commands: view #1 zalign sel view protein zalign sel view sel :<8 zalign sel <https://rbvi.ucsf.edu/chimerax/docs/user/commands/view.html> I hope this helps, Elaine ----- Elaine C. Meng, Ph.D. UCSF Chimera(X) team Department of Pharmaceutical Chemistry University of California, San Francisco
On Feb 5, 2021, at 7:31 AM, Y. Mutum <ym337@cam.ac.uk> wrote:
Hi Elaine
I was thinking of a follow-up on this question, as this question is linked to the previous one.
This answer might be slightly subjective, but given your ChimeraX expertise in visual representation, your thoughts would nonetheless be helpful.
So, now that we have aligned the rotation axis perpendicular to the screen (from the previous question), I am thinking of putting in a small scalebar - say, 50 A. I created this rectange object (command: shape rectange width 50 height 2), along with the rotation axis and the other 3cx5.pdb's in ChimeraX(the email below) • Where would you recommend placing the scale bar (in this case, a rectange) wrt the model 3cx5, considering we want a view perpendicular the rotation axis, (showing the whole protein-complex to get a genral ide of the size of the object)? (considering that a distance is defined for in a plane). • The other related question is - if suppose, we draw a line b/w the Fe-centres of the hemes (by distance command from #1/C:4002 and #2/C:4002), how do we align the rectange object against this distance line. I can think of doing aligning it manually, but this might not be entirely accurate and I am not sure if there's some other method - such as aligning this rectange perpendicular to the rotation-axis line (generated from the previous email).
I am also open to consider other alternatives, rather than this method as well.
Thanks Yaikhomba From: Elaine Meng <meng@cgl.ucsf.edu> Sent: 18 January 2021 18:09 To: Y. Mutum <ym337@cam.ac.uk> Cc: chimerax-users@cgl.ucsf.edu <chimerax-users@cgl.ucsf.edu> Subject: Re: [chimerax-users] Identifying symmetry of a model
Hi Yaikhomba, ChimeraX doesn't have anything to automatically identify the symmetry of an atomic structure. There is only "measure symmetry" for maps, which is also somewhat limited in assuming standard orientations, etc. <https://rbvi.ucsf.edu/chimerax/docs/user/commands/measure.html#symmetry>
However, say you have identified the approximate symmetry by eye. One possibility for C2 symetry is to open two copies of the structure, and then superimpose so that one half in one copy is on top of the other half of the other copy, then measure the rotation between the two. However, if the symmetry is only approximate, it is your own judgment call of which atoms are best to match to which atoms to obtain the best superposition of the halves.
Example: open 3cx5 open 3cx5 mm #1/D to #2/O measure rotation #1 to #2
... the measurement shows an axis which is really two markers (fake atoms) connected by a link (bond). You can manually orient the view to put that axis along the line of sight. You may need to temporarily hide the atomic models (e.g. with checkboxes in the Models panel) to see it clearly. Then you can hide the axis model if you want and turn the atomic models display back on.
Chimera had an "align" command to put two atoms or markers along the line of sight, but ChimeraX does not have that feature. (ChimeraX "align" does something different, it superimposes atoms. You could use it to do the superposition, or use "mm" aka "matchmaker" as in my example above)
<https://rbvi.ucsf.edu/chimerax/docs/user/commands/matchmaker.html> <https://rbvi.ucsf.edu/chimerax/docs/user/commands/measure.html#rotation>
For your 4 residues case, you could define a plane from those residues (displayed as a flat disc) with "define" and then (again manually) rotate the view so that the plane is perpendicular to the line of sight. You can hide atomic models and/or hide the plane at any step with the Model panel checkboxes, as above.
<https://rbvi.ucsf.edu/chimerax/docs/user/commands/define.html#plane>
I hope this helps, Elaine ----- Elaine C. Meng, Ph.D. UCSF Chimera(X) team Department of Pharmaceutical Chemistry University of California, San Francisco
On Jan 18, 2021, at 9:02 AM, Y. Mutum <ym337@cam.ac.uk> wrote:
Hi
I am working with pdb id: 3cx5. This model is roughly C2 symmetric. Is there a way to identify the symmetry (or rough symmetry of a not-so symmetric model) of a pdb/ mmcif model and orient the symmetry axis to the axis perpendicular to the screen?
On a similar note, say we select a 4 somewhat symmetry-related residues in this model and make 2 intersecting lines, is there a way to align these lines perpendicular to the perpendicular screen axis?
Thanks Yaikhomba
_______________________________________________ ChimeraX-users mailing list ChimeraX-users@cgl.ucsf.edu Manage subscription: https://www.rbvi.ucsf.edu/mailman/listinfo/chimerax-users
participants (2)
-
 Elaine Meng
Elaine Meng -
 Y. Mutum
Y. Mutum