
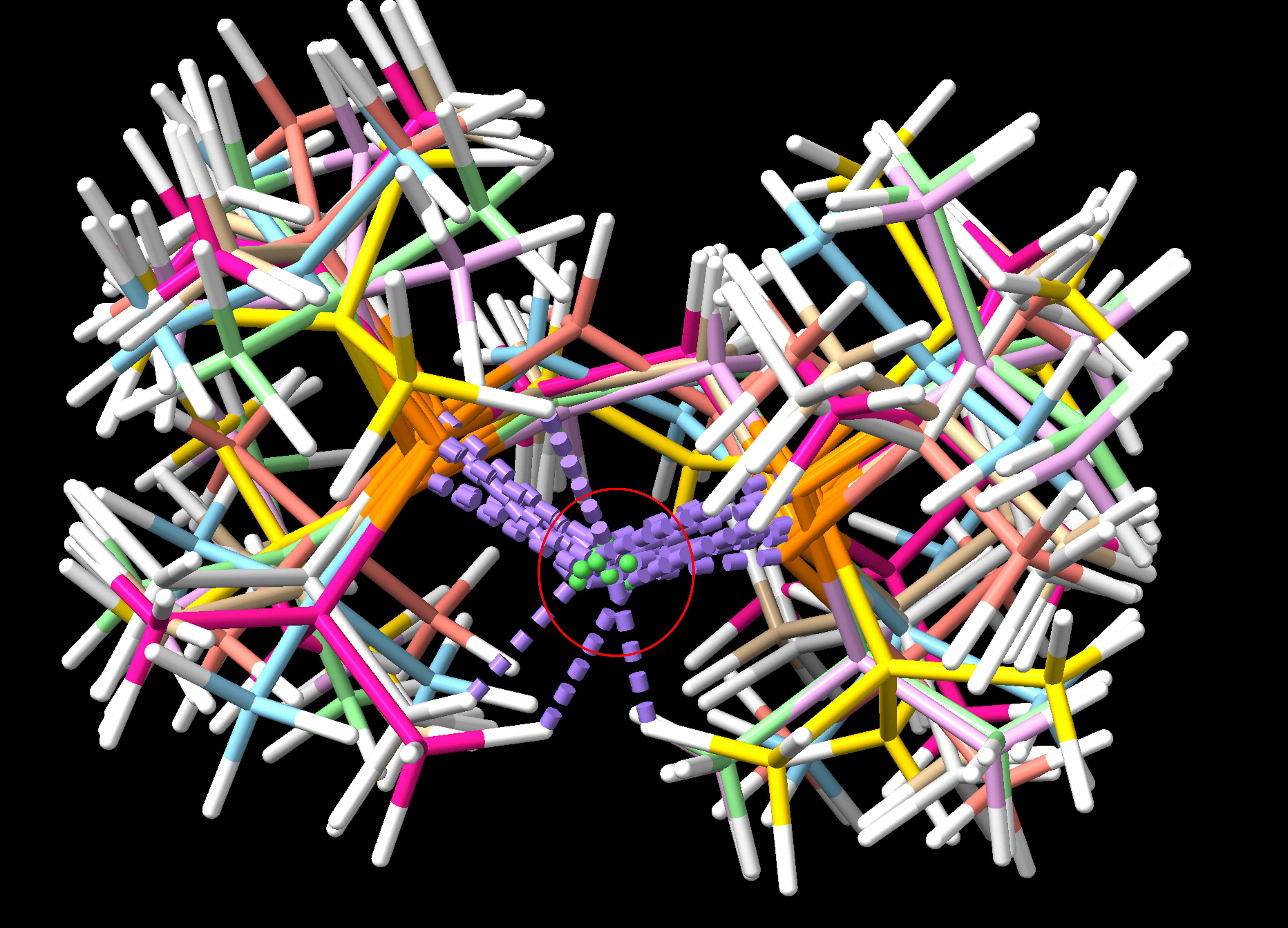
Dear Professor: Hello, I have aligned the atoms #1 of different models with the command "align #1:1 toAtoms #2:1", but I found that the atoms #1 don't overlap very well, which affects the aesthetics of the publication. So, I would like to ask you if there is a way to align the green atoms in the center, thank you very much (see Figure 1)! 化工化工 1366924572@qq.com
{kind=link}

Hello, With "align" you can use whatever atoms you want (not necessarily the whole residue 1), but this requires finding the names of the specific atoms and including them in the command. Then it is a process of trial and error to fit just the atoms that you want and then see if the fit is good enough for you. If not then you might try using a different subset of the atoms. Since you said you want to align the central green atoms, you may want to try only using those green atoms + the two orange atoms coordinating that ion, and at least one other atom (maybe one of the bond partners of the orange atom(s)) from each structure. You would need to know their names in order to write this command, however. I don't know their names in your structure, so this is only an example. You would have to use the atom names in your structure instead. If the green ones were named M and the orange ones R1 and R2 and another bonded to R2 was named C2, something like: align #1:1@m1,r1,r2,c2 to #2:1@m,r1,r2,c2 You can see atom names by putting the mouse over the atom and waiting for the pop-up balloon, and/or by labeling, e.g. label #1:1 atoms I hope this helps, Elaine ----- Elaine C. Meng, Ph.D. UCSF Chimera(X) team Resource for Biocomputing, Visualization, and Informatics Department of Pharmaceutical Chemistry University of California, San Francisco
On Apr 15, 2024, at 4:24 AM, 化工化工 via ChimeraX-users <chimerax-users@cgl.ucsf.edu> wrote:
Dear Professor: Hello, I have aligned the atoms #1 of different models with the command "align #1:1 toAtoms #2:1", but I found that the atoms #1 don't overlap very well, which affects the aesthetics of the publication. So, I would like to ask you if there is a way to align the green atoms in the center, thank you very much (see Figure 1)!
化工化工 1366924572@qq.com
<图片1.png>
Small correction, if the green ones were named M (I accidentally wrote m1 for the first model), it would be something like align #1:1@m,r1,r2,c2 to #2:1@m,r1,r2,c2
On Apr 15, 2024, at 9:17 AM, Elaine Meng via ChimeraX-users <chimerax-users@cgl.ucsf.edu> wrote:
Hello, With "align" you can use whatever atoms you want (not necessarily the whole residue 1), but this requires finding the names of the specific atoms and including them in the command.
Then it is a process of trial and error to fit just the atoms that you want and then see if the fit is good enough for you. If not then you might try using a different subset of the atoms.
Since you said you want to align the central green atoms, you may want to try only using those green atoms + the two orange atoms coordinating that ion, and at least one other atom (maybe one of the bond partners of the orange atom(s)) from each structure. You would need to know their names in order to write this command, however.
I don't know their names in your structure, so this is only an example. You would have to use the atom names in your structure instead. If the green ones were named M and the orange ones R1 and R2 and another bonded to R2 was named C2, something like:
align #1:1@m1,r1,r2,c2 to #2:1@m,r1,r2,c2
You can see atom names by putting the mouse over the atom and waiting for the pop-up balloon, and/or by labeling, e.g.
label #1:1 atoms
I hope this helps, Elaine ----- Elaine C. Meng, Ph.D. UCSF Chimera(X) team Resource for Biocomputing, Visualization, and Informatics Department of Pharmaceutical Chemistry University of California, San Francisco
On Apr 15, 2024, at 4:24 AM, 化工化工 via ChimeraX-users <chimerax-users@cgl.ucsf.edu> wrote:
Dear Professor: Hello, I have aligned the atoms #1 of different models with the command "align #1:1 toAtoms #2:1", but I found that the atoms #1 don't overlap very well, which affects the aesthetics of the publication. So, I would like to ask you if there is a way to align the green atoms in the center, thank you very much (see Figure 1)!
化工化工 1366924572@qq.com
<图片1.png>
_______________________________________________ ChimeraX-users mailing list -- chimerax-users@cgl.ucsf.edu To unsubscribe send an email to chimerax-users-leave@cgl.ucsf.edu Archives: https://mail.cgl.ucsf.edu/mailman/archives/list/chimerax-users@cgl.ucsf.edu/
Thank you very much for your answer, but I will find that there is still no overlap in the metal (green atoms). I wonder if I can use the move command to move all the green atoms of the model to the same Cartesian coordinates to achieve the overlap after alignment? 化工化工 1366924572@qq.com ------------------ 原始邮件 ------------------ 发件人: "chimerax-users" <meng@cgl.ucsf.edu>; 发送时间: 2024年4月16日(星期二) 凌晨0:40 收件人: "化工化工"<1366924572@qq.com>; 抄送: "ChimeraX Users Help"<chimerax-users@cgl.ucsf.edu>; 主题: Re: [chimerax-users] align the atoms Small correction, if the green ones were named M (I accidentally wrote m1 for the first model), it would be something like align #1:1@m,r1,r2,c2 to #2:1@m,r1,r2,c2 > On Apr 15, 2024, at 9:17 AM, Elaine Meng via ChimeraX-users <chimerax-users@cgl.ucsf.edu> wrote: > > Hello, > With "align" you can use whatever atoms you want (not necessarily the whole residue 1), but this requires finding the names of the specific atoms and including them in the command. > > Then it is a process of trial and error to fit just the atoms that you want and then see if the fit is good enough for you. If not then you might try using a different subset of the atoms. > > Since you said you want to align the central green atoms, you may want to try only using those green atoms + the two orange atoms coordinating that ion, and at least one other atom (maybe one of the bond partners of the orange atom(s)) from each structure. You would need to know their names in order to write this command, however. > > I don't know their names in your structure, so this is only an example. You would have to use the atom names in your structure instead. If the green ones were named M and the orange ones R1 and R2 and another bonded to R2 was named C2, something like: > > align #1:1@m1,r1,r2,c2 to #2:1@m,r1,r2,c2 > > You can see atom names by putting the mouse over the atom and waiting for the pop-up balloon, and/or by labeling, e.g. > > label #1:1 atoms > > I hope this helps, > Elaine > ----- > Elaine C. Meng, Ph.D. > UCSF Chimera(X) team > Resource for Biocomputing, Visualization, and Informatics > Department of Pharmaceutical Chemistry > University of California, San Francisco > >> On Apr 15, 2024, at 4:24 AM, 化工化工 via ChimeraX-users <chimerax-users@cgl.ucsf.edu> wrote: >> >> Dear Professor: >> Hello, I have aligned the atoms #1 of different models with the command "align #1:1 toAtoms #2:1", but I found that the atoms #1 don't overlap very well, which affects the aesthetics of the publication. So, I would like to ask you if there is a way to align the green atoms in the center, thank you very much (see Figure 1)! >> >> >> 化工化工 >> 1366924572@qq.com >> >> <图片1.png> > > _______________________________________________ > ChimeraX-users mailing list -- chimerax-users@cgl.ucsf.edu > To unsubscribe send an email to chimerax-users-leave@cgl.ucsf.edu > Archives: https://mail.cgl.ucsf.edu/mailman/archives/list/chimerax-users@cgl.ucsf.edu/ >
Personally, I would try using the "align" command with different sets of atoms a few times first, especially if the structures also need rotational alignment (not just translation). However, if you prefer moving them interactively and judging by eye, you can move the structures one by one manually (with the mouse), which is probably easier than trying to move it with the "move" command. The "move" command just takes the distance you want to move, not the final coordinates of some atom. See the instructions on how you can move structures individually: <https://rbvi.ucsf.edu/chimerax/docs/user/commands/ui.html#selective> I hope this helps, Elaine ----- Elaine C. Meng, Ph.D. UCSF Chimera(X) team Resource for Biocomputing, Visualization, and Informatics Department of Pharmaceutical Chemistry University of California, San Francisco
On Apr 15, 2024, at 7:16 PM, 化工化工 via ChimeraX-users <chimerax-users@cgl.ucsf.edu> wrote:
Thank you very much for your answer, but I will find that there is still no overlap in the metal (green atoms). I wonder if I can use the move command to move all the green atoms of the model to the same Cartesian coordinates to achieve the overlap after alignment?
化工化工 1366924572@qq.com
------------------ 原始邮件 ------------------ 发件人: "chimerax-users" <meng@cgl.ucsf.edu>; 发送时间: 2024年4月16日(星期二) 凌晨0:40 收件人: "化工化工"<1366924572@qq.com>; 抄送: "ChimeraX Users Help"<chimerax-users@cgl.ucsf.edu>; 主题: Re: [chimerax-users] align the atoms
Small correction, if the green ones were named M (I accidentally wrote m1 for the first model), it would be something like
align #1:1@m,r1,r2,c2 to #2:1@m,r1,r2,c2
On Apr 15, 2024, at 9:17 AM, Elaine Meng via ChimeraX-users <chimerax-users@cgl.ucsf.edu> wrote:
Hello, With "align" you can use whatever atoms you want (not necessarily the whole residue 1), but this requires finding the names of the specific atoms and including them in the command.
Then it is a process of trial and error to fit just the atoms that you want and then see if the fit is good enough for you. If not then you might try using a different subset of the atoms.
Since you said you want to align the central green atoms, you may want to try only using those green atoms + the two orange atoms coordinating that ion, and at least one other atom (maybe one of the bond partners of the orange atom(s)) from each structure. You would need to know their names in order to write this command, however.
I don't know their names in your structure, so this is only an example. You would have to use the atom names in your structure instead. If the green ones were named M and the orange ones R1 and R2 and another bonded to R2 was named C2, something like:
align #1:1@m1,r1,r2,c2 to #2:1@m,r1,r2,c2
You can see atom names by putting the mouse over the atom and waiting for the pop-up balloon, and/or by labeling, e.g.
label #1:1 atoms
I hope this helps, Elaine ----- Elaine C. Meng, Ph.D. UCSF Chimera(X) team Resource for Biocomputing, Visualization, and Informatics Department of Pharmaceutical Chemistry University of California, San Francisco
On Apr 15, 2024, at 4:24 AM, 化工化工 via ChimeraX-users <chimerax-users@cgl.ucsf.edu> wrote:
Dear Professor: Hello, I have aligned the atoms #1 of different models with the command "align #1:1 toAtoms #2:1", but I found that the atoms #1 don't overlap very well, which affects the aesthetics of the publication. So, I would like to ask you if there is a way to align the green atoms in the center, thank you very much (see Figure 1)!
化工化工 1366924572@qq.com
<图片1.png>
participants (2)
-
 Elaine Meng
Elaine Meng -
 化工化工
化工化工