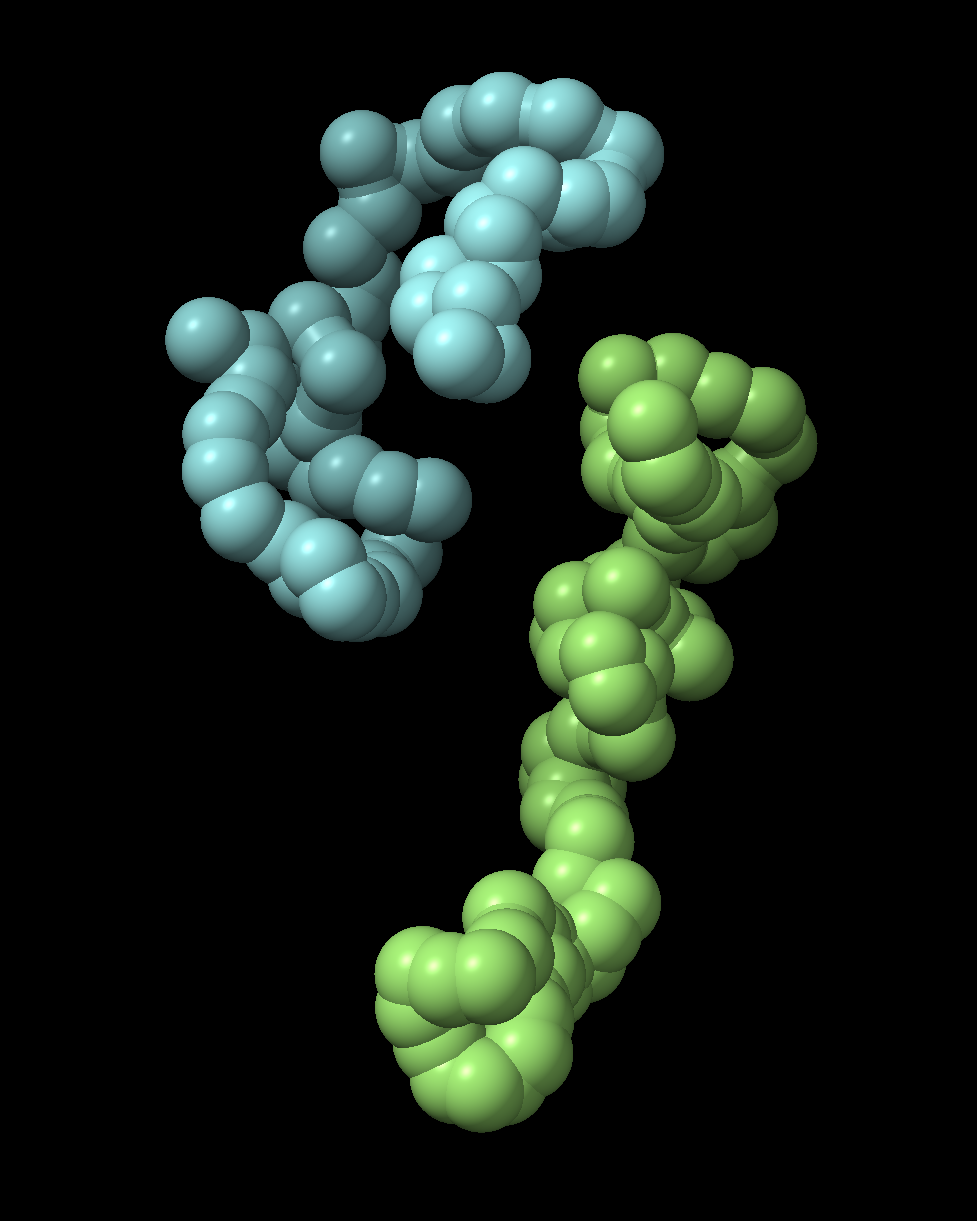
How to Align Chromosome Models with Different Number of Atoms?

Dear ChimeraX User List, How can I compare two chromosome 3D models (PDB files) that have different numbers of "atoms"? Matchmaker tool reports, "Reference and/or match model contains no nucleic or amino acid chains. Use the command-line 'align' command to superimpose small molecules/ligands." align #1 toAtoms #2 reports, "Unequal number of atoms to pair, 49 and 63." align #1 toAtoms #2 matchNumbering true reports, "Pairing dropped 49 atoms and 63 reference atoms. No atoms paired for alignment." align #1:1-40 to #2:1-40 reports, "No atoms paired for alignment." How should I compare two chromosome 3D models below? Thank you. [image: image.png] Best regards, Zichen "Cardiff" Jiang
{kind=link}

Hi Cardiff, It is impossible to tell from a picture what the atoms are named or how the residues are numbered. You have to figure out how to specify an equal number of "atoms" from the two files, based on how the atoms are named and how the residues are numbered. Matchmaker is for biopolymers (proteins or nucleic acids made of real atoms, not your chromosome models), so you would need to use "align" after figuring out how to specify equal numbers of "atoms." You might need to look at your PDB files in a text editor to understand its naming/numbering. Do they really contain 40 residues? Or is each one really a single residue with 40 atoms? "#1:1-40" specifies residues numbered 1-40 in model #1 because the colon symbol ":" means residues. Command "select #1:1-40" will report how many "atoms" that specifies. It might be 0 atoms if the specification is wrong. If you really meant "atoms" numbered 1-40 (not residues) the atom specification should instead be "#1@@serial_number>=1 & #1@@serial_number<=40" How to specify atoms in the command line, where # : @ are symbols for hierarchical levels model residue atom, and @@ refers to atom attributes: <https://rbvi.ucsf.edu/chimerax/docs/user/commands/atomspec.html> I hope this helps, Elaine ----- Elaine C. Meng, Ph.D. UCSF Chimera(X) team Department of Pharmaceutical Chemistry University of California, San Francisco
On Mar 27, 2021, at 11:45 AM, Cardiff Jiang <z8jiang@ucsd.edu> wrote:
Dear ChimeraX User List,
How can I compare two chromosome 3D models (PDB files) that have different numbers of "atoms"?
Matchmaker tool reports, "Reference and/or match model contains no nucleic or amino acid chains. Use the command-line 'align' command to superimpose small molecules/ligands." align #1 toAtoms #2 reports, "Unequal number of atoms to pair, 49 and 63." align #1 toAtoms #2 matchNumbering true reports, "Pairing dropped 49 atoms and 63 reference atoms. No atoms paired for alignment." align #1:1-40 to #2:1-40 reports, "No atoms paired for alignment."
How should I compare two chromosome 3D models below? Thank you. <image.png>
Best regards, Zichen "Cardiff" Jiang _______________________________________________ ChimeraX-users mailing list ChimeraX-users@cgl.ucsf.edu Manage subscription: https://www.rbvi.ucsf.edu/mailman/listinfo/chimerax-users
Dear Elaine, Thank you for your help! The individual balls/ beads of the genome models turn out to be atoms. Is it possible to align four genome models, each with a different number of "atoms"? The smallest atom count is 49 and using the following command sequentially didn't work. The commands aligned model #1 to #2, then separated #1 from #2, finally aligned #1 to #3, and so on: align #1 to #2 #1@@serial_number>=1 & #1@@serial_number<=49 #2@@serial_number>=1 & #2@@serial_number<=49 align #1 to #2 #1@@serial_number>=1 & #1@@serial_number<=49 #3@@serial_number>=1 & #3@@serial_number<=49 Combining the sequential commands together results in "Unequal number of atoms to pair, 49 and 0": align #1 to #2-4 #1@@serial_number>=1 & #1@@serial_number<=49 #2@@serial_number>=1 & #2@@serial_number<= 49 #3@@serial_number>=1 & #3@@serial_number<=49 #4@@serial_number>=1 & #4@@serial_number<= 49 How should I align four models to each other? Thank you. Best regards, Zichen "Cardiff" Jiang On Sat, Mar 27, 2021 at 6:35 PM Elaine Meng <meng@cgl.ucsf.edu> wrote:
Hi Cardiff, It is impossible to tell from a picture what the atoms are named or how the residues are numbered. You have to figure out how to specify an equal number of "atoms" from the two files, based on how the atoms are named and how the residues are numbered. Matchmaker is for biopolymers (proteins or nucleic acids made of real atoms, not your chromosome models), so you would need to use "align" after figuring out how to specify equal numbers of "atoms."
You might need to look at your PDB files in a text editor to understand its naming/numbering. Do they really contain 40 residues? Or is each one really a single residue with 40 atoms?
"#1:1-40" specifies residues numbered 1-40 in model #1 because the colon symbol ":" means residues. Command "select #1:1-40" will report how many "atoms" that specifies. It might be 0 atoms if the specification is wrong. If you really meant "atoms" numbered 1-40 (not residues) the atom specification should instead be "#1@@serial_number>=1 & #1@ @serial_number<=40"
How to specify atoms in the command line, where # : @ are symbols for hierarchical levels model residue atom, and @@ refers to atom attributes: < https://urldefense.com/v3/__https://rbvi.ucsf.edu/chimerax/docs/user/command...
I hope this helps, Elaine ----- Elaine C. Meng, Ph.D. UCSF Chimera(X) team Department of Pharmaceutical Chemistry University of California, San Francisco
On Mar 27, 2021, at 11:45 AM, Cardiff Jiang <z8jiang@ucsd.edu> wrote:
Dear ChimeraX User List,
How can I compare two chromosome 3D models (PDB files) that have different numbers of "atoms"?
Matchmaker tool reports, "Reference and/or match model contains no nucleic or amino acid chains. Use the command-line 'align' command to superimpose small molecules/ligands." align #1 toAtoms #2 reports, "Unequal number of atoms to pair, 49 and 63." align #1 toAtoms #2 matchNumbering true reports, "Pairing dropped 49 atoms and 63 reference atoms. No atoms paired for alignment." align #1:1-40 to #2:1-40 reports, "No atoms paired for alignment."
How should I compare two chromosome 3D models below? Thank you. <image.png>
Best regards, Zichen "Cardiff" Jiang _______________________________________________ ChimeraX-users mailing list ChimeraX-users@cgl.ucsf.edu Manage subscription:
https://urldefense.com/v3/__https://www.rbvi.ucsf.edu/mailman/listinfo/chime...
Hi Cardiff, You can only do pairwise alignments. There is no way to best-fit one model to multiple others at the same time. However, you can still superimpose all of the models together by picking one as the reference (the one that stays in place) and then matching each of the other three to that one. E.g. if #1 is the reference, you could match #2 to #1, then #3 to #1, then #4 to #1. I hope this helps, Elaine ----- Elaine C. Meng, Ph.D. UCSF Chimera(X) team Department of Pharmaceutical Chemistry University of California, San Francisco
On Mar 28, 2021, at 3:17 PM, Cardiff Jiang <z8jiang@ucsd.edu> wrote:
Dear Elaine,
Thank you for your help! The individual balls/ beads of the genome models turn out to be atoms.
Is it possible to align four genome models, each with a different number of "atoms"? The smallest atom count is 49 and using the following command sequentially didn't work. The commands aligned model #1 to #2, then separated #1 from #2, finally aligned #1 to #3, and so on: align #1 to #2 #1@@serial_number>=1 & #1@@serial_number<=49 #2@@serial_number>=1 & #2@@serial_number<=49 align #1 to #2 #1@@serial_number>=1 & #1@@serial_number<=49 #3@@serial_number>=1 & #3@@serial_number<=49
Combining the sequential commands together results in "Unequal number of atoms to pair, 49 and 0": align #1 to #2-4 #1@@serial_number>=1 & #1@@serial_number<=49 #2@@serial_number>=1 & #2@@serial_number<= 49 #3@@serial_number>=1 & #3@@serial_number<=49 #4@@serial_number>=1 & #4@@serial_number<= 49
How should I align four models to each other? Thank you.
Best regards, Zichen "Cardiff" Jiang
On Sat, Mar 27, 2021 at 6:35 PM Elaine Meng <meng@cgl.ucsf.edu> wrote: Hi Cardiff, It is impossible to tell from a picture what the atoms are named or how the residues are numbered. You have to figure out how to specify an equal number of "atoms" from the two files, based on how the atoms are named and how the residues are numbered. Matchmaker is for biopolymers (proteins or nucleic acids made of real atoms, not your chromosome models), so you would need to use "align" after figuring out how to specify equal numbers of "atoms."
You might need to look at your PDB files in a text editor to understand its naming/numbering. Do they really contain 40 residues? Or is each one really a single residue with 40 atoms?
"#1:1-40" specifies residues numbered 1-40 in model #1 because the colon symbol ":" means residues. Command "select #1:1-40" will report how many "atoms" that specifies. It might be 0 atoms if the specification is wrong. If you really meant "atoms" numbered 1-40 (not residues) the atom specification should instead be "#1@@serial_number>=1 & #1@@serial_number<=40"
How to specify atoms in the command line, where # : @ are symbols for hierarchical levels model residue atom, and @@ refers to atom attributes: <https://urldefense.com/v3/__https://rbvi.ucsf.edu/chimerax/docs/user/command... >
I hope this helps, Elaine ----- Elaine C. Meng, Ph.D. UCSF Chimera(X) team Department of Pharmaceutical Chemistry University of California, San Francisco
On Mar 27, 2021, at 11:45 AM, Cardiff Jiang <z8jiang@ucsd.edu> wrote:
Dear ChimeraX User List,
How can I compare two chromosome 3D models (PDB files) that have different numbers of "atoms"?
Matchmaker tool reports, "Reference and/or match model contains no nucleic or amino acid chains. Use the command-line 'align' command to superimpose small molecules/ligands." align #1 toAtoms #2 reports, "Unequal number of atoms to pair, 49 and 63." align #1 toAtoms #2 matchNumbering true reports, "Pairing dropped 49 atoms and 63 reference atoms. No atoms paired for alignment." align #1:1-40 to #2:1-40 reports, "No atoms paired for alignment."
How should I compare two chromosome 3D models below? Thank you. <image.png>
Best regards, Zichen "Cardiff" Jiang _______________________________________________ ChimeraX-users mailing list ChimeraX-users@cgl.ucsf.edu Manage subscription: https://urldefense.com/v3/__https://www.rbvi.ucsf.edu/mailman/listinfo/chime...
Dear Elaine, Just to check if I understood you correctly. I can only superimpose model #1 to #2 in one pairwise alignment job. Then with a separate pairwise alignment job, I can superimpose #1 to #3. I cannot superimpose more than two models simultaneously. Is my understanding correct? Thank you. Best regards, Zichen "Cardiff" Jiang On Sun, Mar 28, 2021 at 6:24 PM Elaine Meng <meng@cgl.ucsf.edu> wrote:
Hi Cardiff, You can only do pairwise alignments. There is no way to best-fit one model to multiple others at the same time.
However, you can still superimpose all of the models together by picking one as the reference (the one that stays in place) and then matching each of the other three to that one. E.g. if #1 is the reference, you could match #2 to #1, then #3 to #1, then #4 to #1.
I hope this helps, Elaine ----- Elaine C. Meng, Ph.D. UCSF Chimera(X) team Department of Pharmaceutical Chemistry University of California, San Francisco
On Mar 28, 2021, at 3:17 PM, Cardiff Jiang <z8jiang@ucsd.edu> wrote:
Dear Elaine,
Thank you for your help! The individual balls/ beads of the genome models turn out to be atoms.
Is it possible to align four genome models, each with a different number of "atoms"? The smallest atom count is 49 and using the following command sequentially didn't work. The commands aligned model #1 to #2, then separated #1 from #2, finally aligned #1 to #3, and so on: align #1 to #2 #1@@serial_number>=1 & #1@@serial_number<=49 #2@@serial_number>=1 & #2@@serial_number<=49 align #1 to #2 #1@@serial_number>=1 & #1@@serial_number<=49 #3@@serial_number>=1 & #3@@serial_number<=49
Combining the sequential commands together results in "Unequal number of atoms to pair, 49 and 0": align #1 to #2-4 #1@@serial_number>=1 & #1@@serial_number<=49 #2@@serial_number>=1 & #2@@serial_number<= 49 #3@@serial_number>=1 & #3@@serial_number<=49 #4@@serial_number>=1 & #4@@serial_number<= 49
How should I align four models to each other? Thank you.
Best regards, Zichen "Cardiff" Jiang
On Sat, Mar 27, 2021 at 6:35 PM Elaine Meng <meng@cgl.ucsf.edu> wrote: Hi Cardiff, It is impossible to tell from a picture what the atoms are named or how the residues are numbered. You have to figure out how to specify an equal number of "atoms" from the two files, based on how the atoms are named and how the residues are numbered. Matchmaker is for biopolymers (proteins or nucleic acids made of real atoms, not your chromosome models), so you would need to use "align" after figuring out how to specify equal numbers of "atoms."
You might need to look at your PDB files in a text editor to understand its naming/numbering. Do they really contain 40 residues? Or is each one really a single residue with 40 atoms?
"#1:1-40" specifies residues numbered 1-40 in model #1 because the colon symbol ":" means residues. Command "select #1:1-40" will report how many "atoms" that specifies. It might be 0 atoms if the specification is wrong. If you really meant "atoms" numbered 1-40 (not residues) the atom specification should instead be "#1@@serial_number>=1 & #1@ @serial_number<=40"
How to specify atoms in the command line, where # : @ are symbols for hierarchical levels model residue atom, and @@ refers to atom attributes: < https://urldefense.com/v3/__https://rbvi.ucsf.edu/chimerax/docs/user/command...
I hope this helps, Elaine ----- Elaine C. Meng, Ph.D. UCSF Chimera(X) team Department of Pharmaceutical Chemistry University of California, San Francisco
On Mar 27, 2021, at 11:45 AM, Cardiff Jiang <z8jiang@ucsd.edu> wrote:
Dear ChimeraX User List,
How can I compare two chromosome 3D models (PDB files) that have different numbers of "atoms"?
Matchmaker tool reports, "Reference and/or match model contains no nucleic or amino acid chains. Use the command-line 'align' command to superimpose small molecules/ligands." align #1 toAtoms #2 reports, "Unequal number of atoms to pair, 49 and 63." align #1 toAtoms #2 matchNumbering true reports, "Pairing dropped 49 atoms and 63 reference atoms. No atoms paired for alignment." align #1:1-40 to #2:1-40 reports, "No atoms paired for alignment."
How should I compare two chromosome 3D models below? Thank you. <image.png>
Best regards, Zichen "Cardiff" Jiang _______________________________________________ ChimeraX-users mailing list ChimeraX-users@cgl.ucsf.edu Manage subscription:
https://urldefense.com/v3/__https://www.rbvi.ucsf.edu/mailman/listinfo/chime...

Ummm ... that won't actually work the way you write it. Be careful about the direction of the superposition. You have to declare one reference protein, and always rotate/translate onto the same coordinate set. So, follow what Elaine wrote exactly. 2 ON 1, then 3 ON 1 etc. For most purposes that is going to give you a good enough solution. But it's not the best one can do, in the same way that a pairwise set of sequence alignments is not going to give you an optimal global alignment. In particular, the details are going to depend on the reference model, so any kind of quantitative analysis is no longer reliable. In general, the problem has conflicting objectives and no principled way to resolve them. If you must, you can use some online tool - PDB eFold at the EBI comes to mind: https://www.ebi.ac.uk/msd-srv/ssm/ But, given all the tradeoffs, the "best" strategy really depends on the exact problem you are trying to solve. TLDR; Multiple sequence alignments, and multiple structure superpositions, are dark arts. :-)
On 2021-03-29, at 11:58, Cardiff Jiang <z8jiang@ucsd.edu> wrote:
EXTERNAL EMAIL: Dear Elaine,
Just to check if I understood you correctly. I can only superimpose model #1 to #2 in one pairwise alignment job. Then with a separate pairwise alignment job, I can superimpose #1 to #3. I cannot superimpose more than two models simultaneously. Is my understanding correct? Thank you.
Best regards, Zichen "Cardiff" Jiang
On Sun, Mar 28, 2021 at 6:24 PM Elaine Meng <meng@cgl.ucsf.edu> wrote: Hi Cardiff, You can only do pairwise alignments. There is no way to best-fit one model to multiple others at the same time.
However, you can still superimpose all of the models together by picking one as the reference (the one that stays in place) and then matching each of the other three to that one. E.g. if #1 is the reference, you could match #2 to #1, then #3 to #1, then #4 to #1.
I hope this helps, Elaine ----- Elaine C. Meng, Ph.D. UCSF Chimera(X) team Department of Pharmaceutical Chemistry University of California, San Francisco
On Mar 28, 2021, at 3:17 PM, Cardiff Jiang <z8jiang@ucsd.edu> wrote:
Dear Elaine,
Thank you for your help! The individual balls/ beads of the genome models turn out to be atoms.
Is it possible to align four genome models, each with a different number of "atoms"? The smallest atom count is 49 and using the following command sequentially didn't work. The commands aligned model #1 to #2, then separated #1 from #2, finally aligned #1 to #3, and so on: align #1 to #2 #1@@serial_number>=1 & #1@@serial_number<=49 #2@@serial_number>=1 & #2@@serial_number<=49 align #1 to #2 #1@@serial_number>=1 & #1@@serial_number<=49 #3@@serial_number>=1 & #3@@serial_number<=49
Combining the sequential commands together results in "Unequal number of atoms to pair, 49 and 0": align #1 to #2-4 #1@@serial_number>=1 & #1@@serial_number<=49 #2@@serial_number>=1 & #2@@serial_number<= 49 #3@@serial_number>=1 & #3@@serial_number<=49 #4@@serial_number>=1 & #4@@serial_number<= 49
How should I align four models to each other? Thank you.
Best regards, Zichen "Cardiff" Jiang
On Sat, Mar 27, 2021 at 6:35 PM Elaine Meng <meng@cgl.ucsf.edu> wrote: Hi Cardiff, It is impossible to tell from a picture what the atoms are named or how the residues are numbered. You have to figure out how to specify an equal number of "atoms" from the two files, based on how the atoms are named and how the residues are numbered. Matchmaker is for biopolymers (proteins or nucleic acids made of real atoms, not your chromosome models), so you would need to use "align" after figuring out how to specify equal numbers of "atoms."
You might need to look at your PDB files in a text editor to understand its naming/numbering. Do they really contain 40 residues? Or is each one really a single residue with 40 atoms?
"#1:1-40" specifies residues numbered 1-40 in model #1 because the colon symbol ":" means residues. Command "select #1:1-40" will report how many "atoms" that specifies. It might be 0 atoms if the specification is wrong. If you really meant "atoms" numbered 1-40 (not residues) the atom specification should instead be "#1@@serial_number>=1 & #1@@serial_number<=40"
How to specify atoms in the command line, where # : @ are symbols for hierarchical levels model residue atom, and @@ refers to atom attributes: <https://urldefense.com/v3/__https://rbvi.ucsf.edu/chimerax/docs/user/command... >
I hope this helps, Elaine ----- Elaine C. Meng, Ph.D. UCSF Chimera(X) team Department of Pharmaceutical Chemistry University of California, San Francisco
On Mar 27, 2021, at 11:45 AM, Cardiff Jiang <z8jiang@ucsd.edu> wrote:
Dear ChimeraX User List,
How can I compare two chromosome 3D models (PDB files) that have different numbers of "atoms"?
Matchmaker tool reports, "Reference and/or match model contains no nucleic or amino acid chains. Use the command-line 'align' command to superimpose small molecules/ligands." align #1 toAtoms #2 reports, "Unequal number of atoms to pair, 49 and 63." align #1 toAtoms #2 matchNumbering true reports, "Pairing dropped 49 atoms and 63 reference atoms. No atoms paired for alignment." align #1:1-40 to #2:1-40 reports, "No atoms paired for alignment."
How should I compare two chromosome 3D models below? Thank you. <image.png>
Best regards, Zichen "Cardiff" Jiang _______________________________________________ ChimeraX-users mailing list ChimeraX-users@cgl.ucsf.edu Manage subscription: https://urldefense.com/v3/__https://www.rbvi.ucsf.edu/mailman/listinfo/chime...
_______________________________________________ ChimeraX-users mailing list ChimeraX-users@cgl.ucsf.edu Manage subscription: https://www.rbvi.ucsf.edu/mailman/listinfo/chimerax-users
Hi Cardiff, It is exactly as Boris said (thanks Boris!) and as I already said in the previous answer: (1) there is no way to best-fit one model to multiple other models at the same time. (2) to superimpose them all together you have to choose one model as the reference, and then for each match, always use that one as the "to" model in the command. Elaine
On Mar 29, 2021, at 12:34 AM, Boris Steipe <boris.steipe@utoronto.ca> wrote:
Ummm ... that won't actually work the way you write it. Be careful about the direction of the superposition. You have to declare one reference protein, and always rotate/translate onto the same coordinate set. So, follow what Elaine wrote exactly. 2 ON 1, then 3 ON 1 etc.
For most purposes that is going to give you a good enough solution. But it's not the best one can do, in the same way that a pairwise set of sequence alignments is not going to give you an optimal global alignment. In particular, the details are going to depend on the reference model, so any kind of quantitative analysis is no longer reliable.
In general, the problem has conflicting objectives and no principled way to resolve them. If you must, you can use some online tool - PDB eFold at the EBI comes to mind: https://www.ebi.ac.uk/msd-srv/ssm/
But, given all the tradeoffs, the "best" strategy really depends on the exact problem you are trying to solve.
TLDR; Multiple sequence alignments, and multiple structure superpositions, are dark arts.
:-)
On 2021-03-29, at 11:58, Cardiff Jiang <z8jiang@ucsd.edu> wrote:
EXTERNAL EMAIL: Dear Elaine,
Just to check if I understood you correctly. I can only superimpose model #1 to #2 in one pairwise alignment job. Then with a separate pairwise alignment job, I can superimpose #1 to #3. I cannot superimpose more than two models simultaneously. Is my understanding correct? Thank you.
Best regards, Zichen "Cardiff" Jiang
On Sun, Mar 28, 2021 at 6:24 PM Elaine Meng <meng@cgl.ucsf.edu> wrote: Hi Cardiff, You can only do pairwise alignments. There is no way to best-fit one model to multiple others at the same time.
However, you can still superimpose all of the models together by picking one as the reference (the one that stays in place) and then matching each of the other three to that one. E.g. if #1 is the reference, you could match #2 to #1, then #3 to #1, then #4 to #1.
I hope this helps, Elaine ----- Elaine C. Meng, Ph.D. UCSF Chimera(X) team Department of Pharmaceutical Chemistry University of California, San Francisco
On Mar 28, 2021, at 3:17 PM, Cardiff Jiang <z8jiang@ucsd.edu> wrote:
Dear Elaine,
Thank you for your help! The individual balls/ beads of the genome models turn out to be atoms.
Is it possible to align four genome models, each with a different number of "atoms"? The smallest atom count is 49 and using the following command sequentially didn't work. The commands aligned model #1 to #2, then separated #1 from #2, finally aligned #1 to #3, and so on: align #1 to #2 #1@@serial_number>=1 & #1@@serial_number<=49 #2@@serial_number>=1 & #2@@serial_number<=49 align #1 to #2 #1@@serial_number>=1 & #1@@serial_number<=49 #3@@serial_number>=1 & #3@@serial_number<=49
Combining the sequential commands together results in "Unequal number of atoms to pair, 49 and 0": align #1 to #2-4 #1@@serial_number>=1 & #1@@serial_number<=49 #2@@serial_number>=1 & #2@@serial_number<= 49 #3@@serial_number>=1 & #3@@serial_number<=49 #4@@serial_number>=1 & #4@@serial_number<= 49
How should I align four models to each other? Thank you.
Best regards, Zichen "Cardiff" Jiang
On Sat, Mar 27, 2021 at 6:35 PM Elaine Meng <meng@cgl.ucsf.edu> wrote: Hi Cardiff, It is impossible to tell from a picture what the atoms are named or how the residues are numbered. You have to figure out how to specify an equal number of "atoms" from the two files, based on how the atoms are named and how the residues are numbered. Matchmaker is for biopolymers (proteins or nucleic acids made of real atoms, not your chromosome models), so you would need to use "align" after figuring out how to specify equal numbers of "atoms."
You might need to look at your PDB files in a text editor to understand its naming/numbering. Do they really contain 40 residues? Or is each one really a single residue with 40 atoms?
"#1:1-40" specifies residues numbered 1-40 in model #1 because the colon symbol ":" means residues. Command "select #1:1-40" will report how many "atoms" that specifies. It might be 0 atoms if the specification is wrong. If you really meant "atoms" numbered 1-40 (not residues) the atom specification should instead be "#1@@serial_number>=1 & #1@@serial_number<=40"
How to specify atoms in the command line, where # : @ are symbols for hierarchical levels model residue atom, and @@ refers to atom attributes: <https://urldefense.com/v3/__https://rbvi.ucsf.edu/chimerax/docs/user/command... >
I hope this helps, Elaine ----- Elaine C. Meng, Ph.D. UCSF Chimera(X) team Department of Pharmaceutical Chemistry University of California, San Francisco
On Mar 27, 2021, at 11:45 AM, Cardiff Jiang <z8jiang@ucsd.edu> wrote:
Dear ChimeraX User List,
How can I compare two chromosome 3D models (PDB files) that have different numbers of "atoms"?
Matchmaker tool reports, "Reference and/or match model contains no nucleic or amino acid chains. Use the command-line 'align' command to superimpose small molecules/ligands." align #1 toAtoms #2 reports, "Unequal number of atoms to pair, 49 and 63." align #1 toAtoms #2 matchNumbering true reports, "Pairing dropped 49 atoms and 63 reference atoms. No atoms paired for alignment." align #1:1-40 to #2:1-40 reports, "No atoms paired for alignment."
How should I compare two chromosome 3D models below? Thank you. <image.png>
Best regards, Zichen "Cardiff" Jiang _______________________________________________ ChimeraX-users mailing list ChimeraX-users@cgl.ucsf.edu Manage subscription: https://urldefense.com/v3/__https://www.rbvi.ucsf.edu/mailman/listinfo/chime...
_______________________________________________ ChimeraX-users mailing list ChimeraX-users@cgl.ucsf.edu Manage subscription: https://www.rbvi.ucsf.edu/mailman/listinfo/chimerax-users
_______________________________________________ ChimeraX-users mailing list ChimeraX-users@cgl.ucsf.edu Manage subscription: https://www.rbvi.ucsf.edu/mailman/listinfo/chimerax-users
Dear Elaine, Unfortunately again, I can't align two models with different numbers of atoms. Please assist. This is the error message: [image: image.png] This is the beginning of the .cmm file for model #1: [image: image.png] This is the beginning of the .cmm file for model #3: [image: image.png] Thank you! Best regards, Zichen “Cardiff” Jiang Undergraduate Class of 2023 | Biology with a Specialization in Bioinformatics Division of Biological Sciences University of California, San Diego On Mon, Mar 29, 2021 at 8:43 AM Elaine Meng <meng@cgl.ucsf.edu> wrote:
Hi Cardiff, It is exactly as Boris said (thanks Boris!) and as I already said in the previous answer: (1) there is no way to best-fit one model to multiple other models at the same time. (2) to superimpose them all together you have to choose one model as the reference, and then for each match, always use that one as the "to" model in the command.
Elaine
On Mar 29, 2021, at 12:34 AM, Boris Steipe <boris.steipe@utoronto.ca> wrote:
Ummm ... that won't actually work the way you write it. Be careful about the direction of the superposition. You have to declare one reference protein, and always rotate/translate onto the same coordinate set. So, follow what Elaine wrote exactly. 2 ON 1, then 3 ON 1 etc.
For most purposes that is going to give you a good enough solution. But it's not the best one can do, in the same way that a pairwise set of sequence alignments is not going to give you an optimal global alignment. In particular, the details are going to depend on the reference model, so any kind of quantitative analysis is no longer reliable.
In general, the problem has conflicting objectives and no principled way to resolve them. If you must, you can use some online tool - PDB eFold at the EBI comes to mind: https://urldefense.com/v3/__https://www.ebi.ac.uk/msd-srv/ssm/__;!!Mih3wA!VD...
But, given all the tradeoffs, the "best" strategy really depends on the exact problem you are trying to solve.
TLDR; Multiple sequence alignments, and multiple structure superpositions, are dark arts.
:-)
On 2021-03-29, at 11:58, Cardiff Jiang <z8jiang@ucsd.edu> wrote:
EXTERNAL EMAIL: Dear Elaine,
Just to check if I understood you correctly. I can only superimpose model #1 to #2 in one pairwise alignment job. Then with a separate pairwise alignment job, I can superimpose #1 to #3. I cannot superimpose more than two models simultaneously. Is my understanding correct? Thank you.
Best regards, Zichen "Cardiff" Jiang
On Sun, Mar 28, 2021 at 6:24 PM Elaine Meng <meng@cgl.ucsf.edu> wrote: Hi Cardiff, You can only do pairwise alignments. There is no way to best-fit one model to multiple others at the same time.
However, you can still superimpose all of the models together by picking one as the reference (the one that stays in place) and then matching each of the other three to that one. E.g. if #1 is the reference, you could match #2 to #1, then #3 to #1, then #4 to #1.
I hope this helps, Elaine ----- Elaine C. Meng, Ph.D. UCSF Chimera(X) team Department of Pharmaceutical Chemistry University of California, San Francisco
On Mar 28, 2021, at 3:17 PM, Cardiff Jiang <z8jiang@ucsd.edu> wrote:
Dear Elaine,
Thank you for your help! The individual balls/ beads of the genome models turn out to be atoms.
Is it possible to align four genome models, each with a different number of "atoms"? The smallest atom count is 49 and using the following command sequentially didn't work. The commands aligned model #1 to #2, then separated #1 from #2, finally aligned #1 to #3, and so on: align #1 to #2 #1@@serial_number>=1 & #1@@serial_number<=49 #2@@serial_number>=1 & #2@@serial_number<=49 align #1 to #2 #1@@serial_number>=1 & #1@@serial_number<=49 #3@@serial_number>=1 & #3@@serial_number<=49
Combining the sequential commands together results in "Unequal number of atoms to pair, 49 and 0": align #1 to #2-4 #1@@serial_number>=1 & #1@@serial_number<=49 #2@@serial_number>=1 & #2@@serial_number<= 49 #3@@serial_number>=1 & #3@@serial_number<=49 #4@@serial_number>=1 & #4@@serial_number<= 49
How should I align four models to each other? Thank you.
Best regards, Zichen "Cardiff" Jiang
On Sat, Mar 27, 2021 at 6:35 PM Elaine Meng <meng@cgl.ucsf.edu> wrote: Hi Cardiff, It is impossible to tell from a picture what the atoms are named or how the residues are numbered. You have to figure out how to specify an equal number of "atoms" from the two files, based on how the atoms are named and how the residues are numbered. Matchmaker is for biopolymers (proteins or nucleic acids made of real atoms, not your chromosome models), so you would need to use "align" after figuring out how to specify equal numbers of "atoms."
You might need to look at your PDB files in a text editor to understand its naming/numbering. Do they really contain 40 residues? Or is each one really a single residue with 40 atoms?
"#1:1-40" specifies residues numbered 1-40 in model #1 because the colon symbol ":" means residues. Command "select #1:1-40" will report how many "atoms" that specifies. It might be 0 atoms if the specification is wrong. If you really meant "atoms" numbered 1-40 (not residues) the atom specification should instead be "#1@@serial_number>=1 & #1@ @serial_number<=40"
How to specify atoms in the command line, where # : @ are symbols for hierarchical levels model residue atom, and @@ refers to atom attributes: < https://urldefense.com/v3/__https://rbvi.ucsf.edu/chimerax/docs/user/command...
I hope this helps, Elaine ----- Elaine C. Meng, Ph.D. UCSF Chimera(X) team Department of Pharmaceutical Chemistry University of California, San Francisco
On Mar 27, 2021, at 11:45 AM, Cardiff Jiang <z8jiang@ucsd.edu> wrote:
Dear ChimeraX User List,
How can I compare two chromosome 3D models (PDB files) that have
different numbers of "atoms"?
Matchmaker tool reports, "Reference and/or match model contains no
nucleic or amino acid chains. Use the command-line 'align' command to superimpose small molecules/ligands." align #1 toAtoms #2 reports, "Unequal number of atoms to pair, 49 and 63." align #1 toAtoms #2 matchNumbering true reports, "Pairing dropped 49 atoms and 63 reference atoms. No atoms paired for alignment." align #1:1-40 to #2:1-40 reports, "No atoms paired for alignment."
How should I compare two chromosome 3D models below? Thank you. <image.png>
Best regards, Zichen "Cardiff" Jiang _______________________________________________ ChimeraX-users mailing list ChimeraX-users@cgl.ucsf.edu Manage subscription:
https://urldefense.com/v3/__https://www.rbvi.ucsf.edu/mailman/listinfo/chime...
_______________________________________________ ChimeraX-users mailing list ChimeraX-users@cgl.ucsf.edu Manage subscription:
https://urldefense.com/v3/__https://www.rbvi.ucsf.edu/mailman/listinfo/chime...
_______________________________________________ ChimeraX-users mailing list ChimeraX-users@cgl.ucsf.edu Manage subscription:
https://urldefense.com/v3/__https://www.rbvi.ucsf.edu/mailman/listinfo/chime...
{kind=link}
{kind=link}
{kind=link}

The marker serial_number attribute in ChimeraX daily builds is the marker's id number which also equals its residue number. Your files show id numbers like 2240 and 4834, nothing in the range of 1 to 73. So of course selecting the serial range 1-73 won't specify anything. So if your markers in #1 have ids 2240 to 2312 (73 total) and in #2 have ids 4834 to 4908 (75 total) and you wanted to exclude the last two markers of #2 in the alignment then use command align #1 to #3:4834-4906 Tom
On Jul 30, 2021, at 9:52 AM, Cardiff Jiang via ChimeraX-users <chimerax-users@cgl.ucsf.edu> wrote:
Dear Elaine,
Unfortunately again, I can't align two models with different numbers of atoms. Please assist.
This is the error message:
This is the beginning of the .cmm file for model #1:
This is the beginning of the .cmm file for model #3:
Thank you!
Best regards,
Zichen “Cardiff” Jiang Undergraduate Class of 2023 | Biology with a Specialization in Bioinformatics Division of Biological Sciences University of California, San Diego
On Mon, Mar 29, 2021 at 8:43 AM Elaine Meng <meng@cgl.ucsf.edu <mailto:meng@cgl.ucsf.edu>> wrote: Hi Cardiff, It is exactly as Boris said (thanks Boris!) and as I already said in the previous answer: (1) there is no way to best-fit one model to multiple other models at the same time. (2) to superimpose them all together you have to choose one model as the reference, and then for each match, always use that one as the "to" model in the command.
Elaine
On Mar 29, 2021, at 12:34 AM, Boris Steipe <boris.steipe@utoronto.ca <mailto:boris.steipe@utoronto.ca>> wrote:
Ummm ... that won't actually work the way you write it. Be careful about the direction of the superposition. You have to declare one reference protein, and always rotate/translate onto the same coordinate set. So, follow what Elaine wrote exactly. 2 ON 1, then 3 ON 1 etc.
For most purposes that is going to give you a good enough solution. But it's not the best one can do, in the same way that a pairwise set of sequence alignments is not going to give you an optimal global alignment. In particular, the details are going to depend on the reference model, so any kind of quantitative analysis is no longer reliable.
In general, the problem has conflicting objectives and no principled way to resolve them. If you must, you can use some online tool - PDB eFold at the EBI comes to mind: https://urldefense.com/v3/__https://www.ebi.ac.uk/msd-srv/ssm/__;!!Mih3wA!VD... <https://urldefense.com/v3/__https://www.ebi.ac.uk/msd-srv/ssm/__;!!Mih3wA!VD...>
But, given all the tradeoffs, the "best" strategy really depends on the exact problem you are trying to solve.
TLDR; Multiple sequence alignments, and multiple structure superpositions, are dark arts.
:-)
On 2021-03-29, at 11:58, Cardiff Jiang <z8jiang@ucsd.edu <mailto:z8jiang@ucsd.edu>> wrote:
EXTERNAL EMAIL: Dear Elaine,
Just to check if I understood you correctly. I can only superimpose model #1 to #2 in one pairwise alignment job. Then with a separate pairwise alignment job, I can superimpose #1 to #3. I cannot superimpose more than two models simultaneously. Is my understanding correct? Thank you.
Best regards, Zichen "Cardiff" Jiang
On Sun, Mar 28, 2021 at 6:24 PM Elaine Meng <meng@cgl.ucsf.edu <mailto:meng@cgl.ucsf.edu>> wrote: Hi Cardiff, You can only do pairwise alignments. There is no way to best-fit one model to multiple others at the same time.
However, you can still superimpose all of the models together by picking one as the reference (the one that stays in place) and then matching each of the other three to that one. E.g. if #1 is the reference, you could match #2 to #1, then #3 to #1, then #4 to #1.
I hope this helps, Elaine ----- Elaine C. Meng, Ph.D. UCSF Chimera(X) team Department of Pharmaceutical Chemistry University of California, San Francisco
On Mar 28, 2021, at 3:17 PM, Cardiff Jiang <z8jiang@ucsd.edu <mailto:z8jiang@ucsd.edu>> wrote:
Dear Elaine,
Thank you for your help! The individual balls/ beads of the genome models turn out to be atoms.
Is it possible to align four genome models, each with a different number of "atoms"? The smallest atom count is 49 and using the following command sequentially didn't work. The commands aligned model #1 to #2, then separated #1 from #2, finally aligned #1 to #3, and so on: align #1 to #2 #1@@serial_number>=1 & #1@@serial_number<=49 #2@@serial_number>=1 & #2@@serial_number<=49 align #1 to #2 #1@@serial_number>=1 & #1@@serial_number<=49 #3@@serial_number>=1 & #3@@serial_number<=49
Combining the sequential commands together results in "Unequal number of atoms to pair, 49 and 0": align #1 to #2-4 #1@@serial_number>=1 & #1@@serial_number<=49 #2@@serial_number>=1 & #2@@serial_number<= 49 #3@@serial_number>=1 & #3@@serial_number<=49 #4@@serial_number>=1 & #4@@serial_number<= 49
How should I align four models to each other? Thank you.
Best regards, Zichen "Cardiff" Jiang
On Sat, Mar 27, 2021 at 6:35 PM Elaine Meng <meng@cgl.ucsf.edu <mailto:meng@cgl.ucsf.edu>> wrote: Hi Cardiff, It is impossible to tell from a picture what the atoms are named or how the residues are numbered. You have to figure out how to specify an equal number of "atoms" from the two files, based on how the atoms are named and how the residues are numbered. Matchmaker is for biopolymers (proteins or nucleic acids made of real atoms, not your chromosome models), so you would need to use "align" after figuring out how to specify equal numbers of "atoms."
You might need to look at your PDB files in a text editor to understand its naming/numbering. Do they really contain 40 residues? Or is each one really a single residue with 40 atoms?
"#1:1-40" specifies residues numbered 1-40 in model #1 because the colon symbol ":" means residues. Command "select #1:1-40" will report how many "atoms" that specifies. It might be 0 atoms if the specification is wrong. If you really meant "atoms" numbered 1-40 (not residues) the atom specification should instead be "#1@@serial_number>=1 & #1@@serial_number<=40"
How to specify atoms in the command line, where # : @ are symbols for hierarchical levels model residue atom, and @@ refers to atom attributes: <https://urldefense.com/v3/__https://rbvi.ucsf.edu/chimerax/docs/user/command... <https://urldefense.com/v3/__https://rbvi.ucsf.edu/chimerax/docs/user/command...> >
I hope this helps, Elaine ----- Elaine C. Meng, Ph.D. UCSF Chimera(X) team Department of Pharmaceutical Chemistry University of California, San Francisco
On Mar 27, 2021, at 11:45 AM, Cardiff Jiang <z8jiang@ucsd.edu <mailto:z8jiang@ucsd.edu>> wrote:
Dear ChimeraX User List,
How can I compare two chromosome 3D models (PDB files) that have different numbers of "atoms"?
Matchmaker tool reports, "Reference and/or match model contains no nucleic or amino acid chains. Use the command-line 'align' command to superimpose small molecules/ligands." align #1 toAtoms #2 reports, "Unequal number of atoms to pair, 49 and 63." align #1 toAtoms #2 matchNumbering true reports, "Pairing dropped 49 atoms and 63 reference atoms. No atoms paired for alignment." align #1:1-40 to #2:1-40 reports, "No atoms paired for alignment."
How should I compare two chromosome 3D models below? Thank you. <image.png>
Best regards, Zichen "Cardiff" Jiang _______________________________________________ ChimeraX-users mailing list ChimeraX-users@cgl.ucsf.edu <mailto:ChimeraX-users@cgl.ucsf.edu> Manage subscription: https://urldefense.com/v3/__https://www.rbvi.ucsf.edu/mailman/listinfo/chime... <https://urldefense.com/v3/__https://www.rbvi.ucsf.edu/mailman/listinfo/chime...>
_______________________________________________ ChimeraX-users mailing list ChimeraX-users@cgl.ucsf.edu <mailto:ChimeraX-users@cgl.ucsf.edu> Manage subscription: https://urldefense.com/v3/__https://www.rbvi.ucsf.edu/mailman/listinfo/chime... <https://urldefense.com/v3/__https://www.rbvi.ucsf.edu/mailman/listinfo/chime...>
_______________________________________________ ChimeraX-users mailing list ChimeraX-users@cgl.ucsf.edu <mailto:ChimeraX-users@cgl.ucsf.edu> Manage subscription: https://urldefense.com/v3/__https://www.rbvi.ucsf.edu/mailman/listinfo/chime... <https://urldefense.com/v3/__https://www.rbvi.ucsf.edu/mailman/listinfo/chime...>
_______________________________________________ ChimeraX-users mailing list ChimeraX-users@cgl.ucsf.edu Manage subscription: https://www.rbvi.ucsf.edu/mailman/listinfo/chimerax-users
{kind=link}
{kind=link}
{kind=link}
Dear Tom, Oops, I understand now. Thank you! Best regards, Zichen “Cardiff” Jiang Undergraduate Class of 2023 | Biology with a Specialization in Bioinformatics Division of Biological Sciences University of California, San Diego On Fri, Jul 30, 2021 at 10:13 AM Tom Goddard <goddard@sonic.net> wrote:
The marker serial_number attribute in ChimeraX daily builds is the marker's id number which also equals its residue number. Your files show id numbers like 2240 and 4834, nothing in the range of 1 to 73. So of course selecting the serial range 1-73 won't specify anything.
So if your markers in #1 have ids 2240 to 2312 (73 total) and in #2 have ids 4834 to 4908 (75 total) and you wanted to exclude the last two markers of #2 in the alignment then use command
align #1 to #3:4834-4906
Tom
On Jul 30, 2021, at 9:52 AM, Cardiff Jiang via ChimeraX-users < chimerax-users@cgl.ucsf.edu> wrote:
Dear Elaine,
Unfortunately again, I can't align two models with different numbers of atoms. Please assist.
This is the error message: [image: image.png]
This is the beginning of the .cmm file for model #1: [image: image.png]
This is the beginning of the .cmm file for model #3: [image: image.png]
Thank you!
Best regards,
Zichen “Cardiff” Jiang Undergraduate Class of 2023 | Biology with a Specialization in Bioinformatics Division of Biological Sciences
University of California, San Diego
On Mon, Mar 29, 2021 at 8:43 AM Elaine Meng <meng@cgl.ucsf.edu> wrote:
Hi Cardiff, It is exactly as Boris said (thanks Boris!) and as I already said in the previous answer: (1) there is no way to best-fit one model to multiple other models at the same time. (2) to superimpose them all together you have to choose one model as the reference, and then for each match, always use that one as the "to" model in the command.
Elaine
On Mar 29, 2021, at 12:34 AM, Boris Steipe <boris.steipe@utoronto.ca> wrote:
Ummm ... that won't actually work the way you write it. Be careful about the direction of the superposition. You have to declare one reference protein, and always rotate/translate onto the same coordinate set. So, follow what Elaine wrote exactly. 2 ON 1, then 3 ON 1 etc.
For most purposes that is going to give you a good enough solution. But it's not the best one can do, in the same way that a pairwise set of sequence alignments is not going to give you an optimal global alignment. In particular, the details are going to depend on the reference model, so any kind of quantitative analysis is no longer reliable.
In general, the problem has conflicting objectives and no principled way to resolve them. If you must, you can use some online tool - PDB eFold at the EBI comes to mind: https://urldefense.com/v3/__https://www.ebi.ac.uk/msd-srv/ssm/__;!!Mih3wA!VD...
But, given all the tradeoffs, the "best" strategy really depends on the exact problem you are trying to solve.
TLDR; Multiple sequence alignments, and multiple structure superpositions, are dark arts.
:-)
On 2021-03-29, at 11:58, Cardiff Jiang <z8jiang@ucsd.edu> wrote:
EXTERNAL EMAIL: Dear Elaine,
Just to check if I understood you correctly. I can only superimpose model #1 to #2 in one pairwise alignment job. Then with a separate pairwise alignment job, I can superimpose #1 to #3. I cannot superimpose more than two models simultaneously. Is my understanding correct? Thank you.
Best regards, Zichen "Cardiff" Jiang
On Sun, Mar 28, 2021 at 6:24 PM Elaine Meng <meng@cgl.ucsf.edu> wrote: Hi Cardiff, You can only do pairwise alignments. There is no way to best-fit one model to multiple others at the same time.
However, you can still superimpose all of the models together by picking one as the reference (the one that stays in place) and then matching each of the other three to that one. E.g. if #1 is the reference, you could match #2 to #1, then #3 to #1, then #4 to #1.
I hope this helps, Elaine ----- Elaine C. Meng, Ph.D. UCSF Chimera(X) team Department of Pharmaceutical Chemistry University of California, San Francisco
On Mar 28, 2021, at 3:17 PM, Cardiff Jiang <z8jiang@ucsd.edu> wrote:
Dear Elaine,
Thank you for your help! The individual balls/ beads of the genome models turn out to be atoms.
Is it possible to align four genome models, each with a different number of "atoms"? The smallest atom count is 49 and using the following command sequentially didn't work. The commands aligned model #1 to #2, then separated #1 from #2, finally aligned #1 to #3, and so on: align #1 to #2 #1@@serial_number>=1 & #1@@serial_number<=49 #2@@serial_number>=1 & #2@@serial_number<=49 align #1 to #2 #1@@serial_number>=1 & #1@@serial_number<=49 #3@@serial_number>=1 & #3@@serial_number<=49
Combining the sequential commands together results in "Unequal number of atoms to pair, 49 and 0": align #1 to #2-4 #1@@serial_number>=1 & #1@@serial_number<=49 #2@@serial_number>=1 & #2@@serial_number<= 49 #3@@serial_number>=1 & #3@@serial_number<=49 #4@@serial_number>=1 & #4@@serial_number<= 49
How should I align four models to each other? Thank you.
Best regards, Zichen "Cardiff" Jiang
On Sat, Mar 27, 2021 at 6:35 PM Elaine Meng <meng@cgl.ucsf.edu> wrote: Hi Cardiff, It is impossible to tell from a picture what the atoms are named or how the residues are numbered. You have to figure out how to specify an equal number of "atoms" from the two files, based on how the atoms are named and how the residues are numbered. Matchmaker is for biopolymers (proteins or nucleic acids made of real atoms, not your chromosome models), so you would need to use "align" after figuring out how to specify equal numbers of "atoms."
You might need to look at your PDB files in a text editor to understand its naming/numbering. Do they really contain 40 residues? Or is each one really a single residue with 40 atoms?
"#1:1-40" specifies residues numbered 1-40 in model #1 because the colon symbol ":" means residues. Command "select #1:1-40" will report how many "atoms" that specifies. It might be 0 atoms if the specification is wrong. If you really meant "atoms" numbered 1-40 (not residues) the atom specification should instead be "#1@@serial_number>=1 & #1@ @serial_number<=40"
How to specify atoms in the command line, where # : @ are symbols for hierarchical levels model residue atom, and @@ refers to atom attributes: < https://urldefense.com/v3/__https://rbvi.ucsf.edu/chimerax/docs/user/command...
I hope this helps, Elaine ----- Elaine C. Meng, Ph.D. UCSF Chimera(X) team Department of Pharmaceutical Chemistry University of California, San Francisco
On Mar 27, 2021, at 11:45 AM, Cardiff Jiang <z8jiang@ucsd.edu>
wrote:
Dear ChimeraX User List,
How can I compare two chromosome 3D models (PDB files) that have
different numbers of "atoms"?
Matchmaker tool reports, "Reference and/or match model contains no
nucleic or amino acid chains. Use the command-line 'align' command to superimpose small molecules/ligands." align #1 toAtoms #2 reports, "Unequal number of atoms to pair, 49 and 63." align #1 toAtoms #2 matchNumbering true reports, "Pairing dropped 49 atoms and 63 reference atoms. No atoms paired for alignment." align #1:1-40 to #2:1-40 reports, "No atoms paired for alignment."
How should I compare two chromosome 3D models below? Thank you. <image.png>
Best regards, Zichen "Cardiff" Jiang _______________________________________________ ChimeraX-users mailing list ChimeraX-users@cgl.ucsf.edu Manage subscription:
https://urldefense.com/v3/__https://www.rbvi.ucsf.edu/mailman/listinfo/chime...
_______________________________________________ ChimeraX-users mailing list ChimeraX-users@cgl.ucsf.edu Manage subscription:
https://urldefense.com/v3/__https://www.rbvi.ucsf.edu/mailman/listinfo/chime...
_______________________________________________ ChimeraX-users mailing list ChimeraX-users@cgl.ucsf.edu Manage subscription:
https://urldefense.com/v3/__https://www.rbvi.ucsf.edu/mailman/listinfo/chime...
_______________________________________________ ChimeraX-users mailing list ChimeraX-users@cgl.ucsf.edu Manage subscription: https://www.rbvi.ucsf.edu/mailman/listinfo/chimerax-users <https://urldefense.proofpoint.com/v2/url?u=https-3A__plato.cgl.ucsf.edu_mail...>
{kind=link}
{kind=link}
{kind=link}
participants (4)
-
 Boris Steipe
Boris Steipe -
 Cardiff Jiang
Cardiff Jiang -
 Elaine Meng
Elaine Meng -
 Tom Goddard
Tom Goddard