
Hello everyone, I'm working to automate making movies from 10,000 frame trajectories. Currently, my cxc contains the following commands windowsize 1920 1080 set bgColor white view zalign ligand inFrontOf #1 sel protein surface; surface color salmon; transparency protein 60 target s sel :UNL color sel dark slate gray; color sel byhetero sel :SIA color sel dim gray; color sel byhetero sel :UNL hbonds :UNL reveal true surface zone #1 nearAtoms ligand distance 15 select zone ligand 15; select add ligand select ~sel ~cartoon sel delete sel ~sel view ligand movie record supersample 4 perframe "~label; label hbondatoms" ; coordset #1 1,10000,50; wait 200; ~perframe movie encode output hbond.mp4 bitrate 6000 framerate 4 The above takes ~ 30 mins, and it's something I would like to do for whole libraries I'm screening. I think the longest process is "hbonds :UNL reveal true" while second longest is movie record and perframe. Is there a way to speed this up? Is there a way to take advantage of multi-threading if it isn't already enabled? If multi-threading is already being used, what can I change to be more efficient? I deleted the parts that I didn't need "delete sel" since they're not involved in the binding site, and I thought it would help speed up surface calculations while recording. Regards, Alex Lee PhD Student Woods Research Group<https://woodsgroup.franklinresearch.uga.edu/> Department of Biochemistry & Molecular Biology Complex Carbohydrate Research Center University of Georgia 315 Riverbend Rd. Athens, GA 30602 Email: ahl63010@uga.edu Mobile: (470) 641-3884

Hi Alex, Yes, the bottlenecks are likely the surface recomputations as the trajectory plays, and the H-bonds calculation for 10,000 frames. I believe the surface calculation automatically uses multiple processors if available, so there's nothing you can do there, and the H-bonds calculation is very complicated and has not been parallelized (nor would it be easy to). My advice would be to use fewer frames. A 10,000 frame trajectory shown at 25 frames/sec would produce a movie close to 7 minutes long. Is that really what you want? If you used every 10th frame ("step 10" in the open command) then the movie would be 40 seconds long and take 3 minutes to record. --Eric Eric Pettersen UCSF Computer Graphics Lab
On Sep 28, 2023, at 5:55 PM, Alexander Hung Lee via ChimeraX-users <chimerax-users@cgl.ucsf.edu> wrote:
Hello everyone,
I'm working to automate making movies from 10,000 frame trajectories. Currently, my cxc contains the following commands
windowsize 1920 1080 set bgColor white view zalign ligand inFrontOf #1 sel protein surface; surface color salmon; transparency protein 60 target s sel :UNL color sel dark slate gray; color sel byhetero sel :SIA color sel dim gray; color sel byhetero sel :UNL hbonds :UNL reveal true surface zone #1 nearAtoms ligand distance 15 select zone ligand 15; select add ligand select ~sel ~cartoon sel delete sel ~sel view ligand movie record supersample 4 perframe "~label; label hbondatoms" ; coordset #1 1,10000,50; wait 200; ~perframe movie encode output hbond.mp4 bitrate 6000 framerate 4
The above takes ~ 30 mins, and it's something I would like to do for whole libraries I'm screening. I think the longest process is "hbonds :UNL reveal true" while second longest is movie record and perframe. Is there a way to speed this up? Is there a way to take advantage of multi-threading if it isn't already enabled? If multi-threading is already being used, what can I change to be more efficient?
I deleted the parts that I didn't need "delete sel" since they're not involved in the binding site, and I thought it would help speed up surface calculations while recording.
Regards, Alex Lee PhD Student Woods Research Group <https://woodsgroup.franklinresearch.uga.edu/> Department of Biochemistry & Molecular Biology Complex Carbohydrate Research Center University of Georgia
315 Riverbend Rd. Athens, GA 30602 Email: ahl63010@uga.edu <mailto:ahl63010@uga.edu> Mobile: (470) 641-3884 _______________________________________________ ChimeraX-users mailing list -- chimerax-users@cgl.ucsf.edu <mailto:chimerax-users@cgl.ucsf.edu> To unsubscribe send an email to chimerax-users-leave@cgl.ucsf.edu <mailto:chimerax-users-leave@cgl.ucsf.edu> Archives: https://mail.cgl.ucsf.edu/mailman/archives/list/chimerax-users@cgl.ucsf.edu/ <https://mail.cgl.ucsf.edu/mailman/archives/list/chimerax-users@cgl.ucsf.edu/>
Hi Eric, Thanks for the clarification. Little disappointed that hbond step can't leverage my hardware, but it's good to know I might be able to go around it by manipulating the frames. I'm using 10,000 frames for 100 ns MD simulation. Currently, my cxc workflow is producing ~ 1 min long movie by playing 10,000 frames in 50 frame increments and encoding at 4 fps. I'm visualizing the trajectory with "open *.pdb" followed by "open *.nc format Amber". If I were to use "open *.nc format Amber step 10" instead, would calculations be faster and I should change coordset #1 1,10000,50; wait 200 > coordset #1 1,1000,5; wait 200 ? Surface and hbond calculations should be significantly shorter (hbonds alone take 20 mins)? Thanks, Alex Lee Woods Lab ahl63010@uga.edu ________________________________ From: Eric Pettersen <pett@cgl.ucsf.edu> Sent: Thursday, September 28, 2023 10:12 PM To: Alexander Hung Lee <Alexander.Lee@uga.edu> Cc: chimerax-users@cgl.ucsf.edu <chimerax-users@cgl.ucsf.edu> Subject: Re: [chimerax-users] Speeding up commands from cxc You don't often get email from pett@cgl.ucsf.edu. Learn why this is important<https://aka.ms/LearnAboutSenderIdentification> [EXTERNAL SENDER - PROCEED CAUTIOUSLY] Hi Alex, Yes, the bottlenecks are likely the surface recomputations as the trajectory plays, and the H-bonds calculation for 10,000 frames. I believe the surface calculation automatically uses multiple processors if available, so there's nothing you can do there, and the H-bonds calculation is very complicated and has not been parallelized (nor would it be easy to). My advice would be to use fewer frames. A 10,000 frame trajectory shown at 25 frames/sec would produce a movie close to 7 minutes long. Is that really what you want? If you used every 10th frame ("step 10" in the open command) then the movie would be 40 seconds long and take 3 minutes to record. --Eric Eric Pettersen UCSF Computer Graphics Lab On Sep 28, 2023, at 5:55 PM, Alexander Hung Lee via ChimeraX-users <chimerax-users@cgl.ucsf.edu<mailto:chimerax-users@cgl.ucsf.edu>> wrote: Hello everyone, I'm working to automate making movies from 10,000 frame trajectories. Currently, my cxc contains the following commands windowsize 1920 1080 set bgColor white view zalign ligand inFrontOf #1 sel protein surface; surface color salmon; transparency protein 60 target s sel :UNL color sel dark slate gray; color sel byhetero sel :SIA color sel dim gray; color sel byhetero sel :UNL hbonds :UNL reveal true surface zone #1 nearAtoms ligand distance 15 select zone ligand 15; select add ligand select ~sel ~cartoon sel delete sel ~sel view ligand movie record supersample 4 perframe "~label; label hbondatoms" ; coordset #1 1,10000,50; wait 200; ~perframe movie encode output hbond.mp4 bitrate 6000 framerate 4 The above takes ~ 30 mins, and it's something I would like to do for whole libraries I'm screening. I think the longest process is "hbonds :UNL reveal true" while second longest is movie record and perframe. Is there a way to speed this up? Is there a way to take advantage of multi-threading if it isn't already enabled? If multi-threading is already being used, what can I change to be more efficient? I deleted the parts that I didn't need "delete sel" since they're not involved in the binding site, and I thought it would help speed up surface calculations while recording. Regards, Alex Lee PhD Student Woods Research Group<https://woodsgroup.franklinresearch.uga.edu/> Department of Biochemistry & Molecular Biology Complex Carbohydrate Research Center University of Georgia 315 Riverbend Rd. Athens, GA 30602 Email: ahl63010@uga.edu<mailto:ahl63010@uga.edu> Mobile: (470) 641-3884 _______________________________________________ ChimeraX-users mailing list -- chimerax-users@cgl.ucsf.edu<mailto:chimerax-users@cgl.ucsf.edu> To unsubscribe send an email to chimerax-users-leave@cgl.ucsf.edu<mailto:chimerax-users-leave@cgl.ucsf.edu> Archives: https://mail.cgl.ucsf.edu/mailman/archives/list/chimerax-users@cgl.ucsf.edu/
Hi Alex, I didn't even notice that you were already only showing every 50th frame. In that case you should use "step 50" in the open command and your play-through command becomes simply "coordset #1 1,200; wait 200". You have to realize that by default when you run the hbonds command on a trajectory model, it computes the H-bonds for every frame of the trajectory (controllable with the "coordsets" keyword). So using a trajectory of 200 frames vs. 10,000 will produce a 50x speedup, so the H-bonds calculation for 200 frames should take on the order of a half minute or so in your case. --Eric
On Sep 28, 2023, at 8:18 PM, Alexander Hung Lee via ChimeraX-users <chimerax-users@cgl.ucsf.edu> wrote:
Hi Eric,
Thanks for the clarification. Little disappointed that hbond step can't leverage my hardware, but it's good to know I might be able to go around it by manipulating the frames. I'm using 10,000 frames for 100 ns MD simulation. Currently, my cxc workflow is producing ~ 1 min long movie by playing 10,000 frames in 50 frame increments and encoding at 4 fps.
I'm visualizing the trajectory with "open *.pdb" followed by "open *.nc format Amber". If I were to use "open *.nc format Amber step 10" instead, would calculations be faster and I should change coordset #1 1,10000,50; wait 200 > coordset #1 1,1000,5; wait 200 ? Surface and hbond calculations should be significantly shorter (hbonds alone take 20 mins)?
Thanks, Alex Lee Woods Lab ahl63010@uga.edu <mailto:ahl63010@uga.edu> From: Eric Pettersen <pett@cgl.ucsf.edu <mailto:pett@cgl.ucsf.edu>> Sent: Thursday, September 28, 2023 10:12 PM To: Alexander Hung Lee <Alexander.Lee@uga.edu <mailto:Alexander.Lee@uga.edu>> Cc: chimerax-users@cgl.ucsf.edu <mailto:chimerax-users@cgl.ucsf.edu> <chimerax-users@cgl.ucsf.edu <mailto:chimerax-users@cgl.ucsf.edu>> Subject: Re: [chimerax-users] Speeding up commands from cxc
You don't often get email from pett@cgl.ucsf.edu <mailto:pett@cgl.ucsf.edu>. Learn why this is important <https://aka.ms/LearnAboutSenderIdentification> [EXTERNAL SENDER - PROCEED CAUTIOUSLY]
Hi Alex, Yes, the bottlenecks are likely the surface recomputations as the trajectory plays, and the H-bonds calculation for 10,000 frames. I believe the surface calculation automatically uses multiple processors if available, so there's nothing you can do there, and the H-bonds calculation is very complicated and has not been parallelized (nor would it be easy to). My advice would be to use fewer frames. A 10,000 frame trajectory shown at 25 frames/sec would produce a movie close to 7 minutes long. Is that really what you want? If you used every 10th frame ("step 10" in the open command) then the movie would be 40 seconds long and take 3 minutes to record.
--Eric
Eric Pettersen UCSF Computer Graphics Lab
On Sep 28, 2023, at 5:55 PM, Alexander Hung Lee via ChimeraX-users <chimerax-users@cgl.ucsf.edu <mailto:chimerax-users@cgl.ucsf.edu>> wrote:
Hello everyone,
I'm working to automate making movies from 10,000 frame trajectories. Currently, my cxc contains the following commands
windowsize 1920 1080 set bgColor white view zalign ligand inFrontOf #1 sel protein surface; surface color salmon; transparency protein 60 target s sel :UNL color sel dark slate gray; color sel byhetero sel :SIA color sel dim gray; color sel byhetero sel :UNL hbonds :UNL reveal true surface zone #1 nearAtoms ligand distance 15 select zone ligand 15; select add ligand select ~sel ~cartoon sel delete sel ~sel view ligand movie record supersample 4 perframe "~label; label hbondatoms" ; coordset #1 1,10000,50; wait 200; ~perframe movie encode output hbond.mp4 bitrate 6000 framerate 4
The above takes ~ 30 mins, and it's something I would like to do for whole libraries I'm screening. I think the longest process is "hbonds :UNL reveal true" while second longest is movie record and perframe. Is there a way to speed this up? Is there a way to take advantage of multi-threading if it isn't already enabled? If multi-threading is already being used, what can I change to be more efficient?
I deleted the parts that I didn't need "delete sel" since they're not involved in the binding site, and I thought it would help speed up surface calculations while recording.
Regards, Alex Lee PhD Student Woods Research Group <https://woodsgroup.franklinresearch.uga.edu/> Department of Biochemistry & Molecular Biology Complex Carbohydrate Research Center University of Georgia
315 Riverbend Rd. Athens, GA 30602 Email: ahl63010@uga.edu <mailto:ahl63010@uga.edu> Mobile: (470) 641-3884 _______________________________________________ ChimeraX-users mailing list -- chimerax-users@cgl.ucsf.edu <mailto:chimerax-users@cgl.ucsf.edu> To unsubscribe send an email to chimerax-users-leave@cgl.ucsf.edu <mailto:chimerax-users-leave@cgl.ucsf.edu> Archives: https://mail.cgl.ucsf.edu/mailman/archives/list/chimerax-users@cgl.ucsf.edu/ <https://mail.cgl.ucsf.edu/mailman/archives/list/chimerax-users@cgl.ucsf.edu/>
ChimeraX-users mailing list -- chimerax-users@cgl.ucsf.edu To unsubscribe send an email to chimerax-users-leave@cgl.ucsf.edu Archives: https://mail.cgl.ucsf.edu/mailman/archives/list/chimerax-users@cgl.ucsf.edu/
Hi Eric, I've done the following commands on 3 .pdb files and used matchmaker on the 200th frame and 10,000th frame, respectively. open */md.nc structureModel #1 step 50 (slider shows 200 frames loaded) I've also tried open */md.nc structureModel #1 start 1 end 10000 step 50 (same result as above) vs open */md.nc structureModel #2 (slider shows 10,000 frames loaded - original) Both the 200/200 frames versions match the 200/10,000 frames version. If it worked, would 200/200 frames match 10,000/10,000 frames? Alex Lee Woods Lab ahl63010@uga.edu ________________________________ From: Eric Pettersen <pett@cgl.ucsf.edu> Sent: Friday, September 29, 2023 4:54 PM To: Alexander Hung Lee <Alexander.Lee@uga.edu> Cc: ChimeraX Users Help <chimerax-users@cgl.ucsf.edu> Subject: Re: [chimerax-users] Speeding up commands from cxc You don't often get email from pett@cgl.ucsf.edu. Learn why this is important<https://aka.ms/LearnAboutSenderIdentification> [EXTERNAL SENDER - PROCEED CAUTIOUSLY] Hi Alex, I didn't even notice that you were already only showing every 50th frame. In that case you should use "step 50" in the open command and your play-through command becomes simply "coordset #1 1,200; wait 200". You have to realize that by default when you run the hbonds command on a trajectory model, it computes the H-bonds for every frame of the trajectory (controllable with the "coordsets" keyword). So using a trajectory of 200 frames vs. 10,000 will produce a 50x speedup, so the H-bonds calculation for 200 frames should take on the order of a half minute or so in your case. --Eric On Sep 28, 2023, at 8:18 PM, Alexander Hung Lee via ChimeraX-users <chimerax-users@cgl.ucsf.edu<mailto:chimerax-users@cgl.ucsf.edu>> wrote: Hi Eric, Thanks for the clarification. Little disappointed that hbond step can't leverage my hardware, but it's good to know I might be able to go around it by manipulating the frames. I'm using 10,000 frames for 100 ns MD simulation. Currently, my cxc workflow is producing ~ 1 min long movie by playing 10,000 frames in 50 frame increments and encoding at 4 fps. I'm visualizing the trajectory with "open *.pdb" followed by "open *.nc format Amber". If I were to use "open *.nc format Amber step 10" instead, would calculations be faster and I should change coordset #1 1,10000,50; wait 200 > coordset #1 1,1000,5; wait 200 ? Surface and hbond calculations should be significantly shorter (hbonds alone take 20 mins)? Thanks, Alex Lee Woods Lab ahl63010@uga.edu<mailto:ahl63010@uga.edu> ________________________________ From: Eric Pettersen <pett@cgl.ucsf.edu<mailto:pett@cgl.ucsf.edu>> Sent: Thursday, September 28, 2023 10:12 PM To: Alexander Hung Lee <Alexander.Lee@uga.edu<mailto:Alexander.Lee@uga.edu>> Cc: chimerax-users@cgl.ucsf.edu<mailto:chimerax-users@cgl.ucsf.edu> <chimerax-users@cgl.ucsf.edu<mailto:chimerax-users@cgl.ucsf.edu>> Subject: Re: [chimerax-users] Speeding up commands from cxc You don't often get email from pett@cgl.ucsf.edu<mailto:pett@cgl.ucsf.edu>. Learn why this is important<https://aka.ms/LearnAboutSenderIdentification> [EXTERNAL SENDER - PROCEED CAUTIOUSLY] Hi Alex, Yes, the bottlenecks are likely the surface recomputations as the trajectory plays, and the H-bonds calculation for 10,000 frames. I believe the surface calculation automatically uses multiple processors if available, so there's nothing you can do there, and the H-bonds calculation is very complicated and has not been parallelized (nor would it be easy to). My advice would be to use fewer frames. A 10,000 frame trajectory shown at 25 frames/sec would produce a movie close to 7 minutes long. Is that really what you want? If you used every 10th frame ("step 10" in the open command) then the movie would be 40 seconds long and take 3 minutes to record. --Eric Eric Pettersen UCSF Computer Graphics Lab On Sep 28, 2023, at 5:55 PM, Alexander Hung Lee via ChimeraX-users <chimerax-users@cgl.ucsf.edu<mailto:chimerax-users@cgl.ucsf.edu>> wrote: Hello everyone, I'm working to automate making movies from 10,000 frame trajectories. Currently, my cxc contains the following commands windowsize 1920 1080 set bgColor white view zalign ligand inFrontOf #1 sel protein surface; surface color salmon; transparency protein 60 target s sel :UNL color sel dark slate gray; color sel byhetero sel :SIA color sel dim gray; color sel byhetero sel :UNL hbonds :UNL reveal true surface zone #1 nearAtoms ligand distance 15 select zone ligand 15; select add ligand select ~sel ~cartoon sel delete sel ~sel view ligand movie record supersample 4 perframe "~label; label hbondatoms" ; coordset #1 1,10000,50; wait 200; ~perframe movie encode output hbond.mp4 bitrate 6000 framerate 4 The above takes ~ 30 mins, and it's something I would like to do for whole libraries I'm screening. I think the longest process is "hbonds :UNL reveal true" while second longest is movie record and perframe. Is there a way to speed this up? Is there a way to take advantage of multi-threading if it isn't already enabled? If multi-threading is already being used, what can I change to be more efficient? I deleted the parts that I didn't need "delete sel" since they're not involved in the binding site, and I thought it would help speed up surface calculations while recording. Regards, Alex Lee PhD Student Woods Research Group<https://woodsgroup.franklinresearch.uga.edu/> Department of Biochemistry & Molecular Biology Complex Carbohydrate Research Center University of Georgia 315 Riverbend Rd. Athens, GA 30602 Email: ahl63010@uga.edu<mailto:ahl63010@uga.edu> Mobile: (470) 641-3884 _______________________________________________ ChimeraX-users mailing list -- chimerax-users@cgl.ucsf.edu<mailto:chimerax-users@cgl.ucsf.edu> To unsubscribe send an email to chimerax-users-leave@cgl.ucsf.edu<mailto:chimerax-users-leave@cgl.ucsf.edu> Archives: https://mail.cgl.ucsf.edu/mailman/archives/list/chimerax-users@cgl.ucsf.edu/ _______________________________________________ ChimeraX-users mailing list -- chimerax-users@cgl.ucsf.edu<mailto:chimerax-users@cgl.ucsf.edu> To unsubscribe send an email to chimerax-users-leave@cgl.ucsf.edu<mailto:chimerax-users-leave@cgl.ucsf.edu> Archives: https://mail.cgl.ucsf.edu/mailman/archives/list/chimerax-users@cgl.ucsf.edu/
Hi Alex, If I’m understanding the question correctly, no, frame 200 will not match frame 10,000. The first two commands you provided use the following frames from the overall trajectory: 1, 51, 101, … 9901, 9951, so the first frames will match, but the last frames will not. If you wanted the last frames to match then you would have to specify “start 50”, which case the 200 frames would be 50, 100, 150, … 9950, 10000. —Eric
On Sep 30, 2023, at 7:19 PM, Alexander Hung Lee via ChimeraX-users <chimerax-users@cgl.ucsf.edu> wrote:
Hi Eric,
I've done the following commands on 3 .pdb files and used matchmaker on the 200th frame and 10,000th frame, respectively.
open */md.nc structureModel #1 step 50 (slider shows 200 frames loaded) I've also tried open */md.nc structureModel #1 start 1 end 10000 step 50 (same result as above)
vs open */md.nc structureModel #2 (slider shows 10,000 frames loaded - original)
Both the 200/200 frames versions match the 200/10,000 frames version. If it worked, would 200/200 frames match 10,000/10,000 frames?
Alex Lee Woods Lab ahl63010@uga.edu <mailto:ahl63010@uga.edu> From: Eric Pettersen <pett@cgl.ucsf.edu <mailto:pett@cgl.ucsf.edu>> Sent: Friday, September 29, 2023 4:54 PM To: Alexander Hung Lee <Alexander.Lee@uga.edu <mailto:Alexander.Lee@uga.edu>> Cc: ChimeraX Users Help <chimerax-users@cgl.ucsf.edu <mailto:chimerax-users@cgl.ucsf.edu>> Subject: Re: [chimerax-users] Speeding up commands from cxc
You don't often get email from pett@cgl.ucsf.edu <mailto:pett@cgl.ucsf.edu>. Learn why this is important <https://aka.ms/LearnAboutSenderIdentification> [EXTERNAL SENDER - PROCEED CAUTIOUSLY]
Hi Alex, I didn't even notice that you were already only showing every 50th frame. In that case you should use "step 50" in the open command and your play-through command becomes simply "coordset #1 1,200; wait 200". You have to realize that by default when you run the hbonds command on a trajectory model, it computes the H-bonds for every frame of the trajectory (controllable with the "coordsets" keyword). So using a trajectory of 200 frames vs. 10,000 will produce a 50x speedup, so the H-bonds calculation for 200 frames should take on the order of a half minute or so in your case.
--Eric
On Sep 28, 2023, at 8:18 PM, Alexander Hung Lee via ChimeraX-users <chimerax-users@cgl.ucsf.edu <mailto:chimerax-users@cgl.ucsf.edu>> wrote:
Hi Eric,
Thanks for the clarification. Little disappointed that hbond step can't leverage my hardware, but it's good to know I might be able to go around it by manipulating the frames. I'm using 10,000 frames for 100 ns MD simulation. Currently, my cxc workflow is producing ~ 1 min long movie by playing 10,000 frames in 50 frame increments and encoding at 4 fps.
I'm visualizing the trajectory with "open *.pdb" followed by "open *.nc format Amber". If I were to use "open *.nc format Amber step 10" instead, would calculations be faster and I should change coordset #1 1,10000,50; wait 200 > coordset #1 1,1000,5; wait 200 ? Surface and hbond calculations should be significantly shorter (hbonds alone take 20 mins)?
Thanks, Alex Lee Woods Lab ahl63010@uga.edu <mailto:ahl63010@uga.edu> From: Eric Pettersen <pett@cgl.ucsf.edu <mailto:pett@cgl.ucsf.edu>> Sent: Thursday, September 28, 2023 10:12 PM To: Alexander Hung Lee <Alexander.Lee@uga.edu <mailto:Alexander.Lee@uga.edu>> Cc: chimerax-users@cgl.ucsf.edu <mailto:chimerax-users@cgl.ucsf.edu> <chimerax-users@cgl.ucsf.edu <mailto:chimerax-users@cgl.ucsf.edu>> Subject: Re: [chimerax-users] Speeding up commands from cxc
You don't often get email from pett@cgl.ucsf.edu <mailto:pett@cgl.ucsf.edu>. Learn why this is important <https://aka.ms/LearnAboutSenderIdentification> [EXTERNAL SENDER - PROCEED CAUTIOUSLY]
Hi Alex, Yes, the bottlenecks are likely the surface recomputations as the trajectory plays, and the H-bonds calculation for 10,000 frames. I believe the surface calculation automatically uses multiple processors if available, so there's nothing you can do there, and the H-bonds calculation is very complicated and has not been parallelized (nor would it be easy to). My advice would be to use fewer frames. A 10,000 frame trajectory shown at 25 frames/sec would produce a movie close to 7 minutes long. Is that really what you want? If you used every 10th frame ("step 10" in the open command) then the movie would be 40 seconds long and take 3 minutes to record.
--Eric
Eric Pettersen UCSF Computer Graphics Lab
On Sep 28, 2023, at 5:55 PM, Alexander Hung Lee via ChimeraX-users <chimerax-users@cgl.ucsf.edu <mailto:chimerax-users@cgl.ucsf.edu>> wrote:
Hello everyone,
I'm working to automate making movies from 10,000 frame trajectories. Currently, my cxc contains the following commands
windowsize 1920 1080 set bgColor white view zalign ligand inFrontOf #1 sel protein surface; surface color salmon; transparency protein 60 target s sel :UNL color sel dark slate gray; color sel byhetero sel :SIA color sel dim gray; color sel byhetero sel :UNL hbonds :UNL reveal true surface zone #1 nearAtoms ligand distance 15 select zone ligand 15; select add ligand select ~sel ~cartoon sel delete sel ~sel view ligand movie record supersample 4 perframe "~label; label hbondatoms" ; coordset #1 1,10000,50; wait 200; ~perframe movie encode output hbond.mp4 bitrate 6000 framerate 4
The above takes ~ 30 mins, and it's something I would like to do for whole libraries I'm screening. I think the longest process is "hbonds :UNL reveal true" while second longest is movie record and perframe. Is there a way to speed this up? Is there a way to take advantage of multi-threading if it isn't already enabled? If multi-threading is already being used, what can I change to be more efficient?
I deleted the parts that I didn't need "delete sel" since they're not involved in the binding site, and I thought it would help speed up surface calculations while recording.
Regards, Alex Lee PhD Student Woods Research Group <https://woodsgroup.franklinresearch.uga.edu/> Department of Biochemistry & Molecular Biology Complex Carbohydrate Research Center University of Georgia
315 Riverbend Rd. Athens, GA 30602 Email: ahl63010@uga.edu <mailto:ahl63010@uga.edu> Mobile: (470) 641-3884 _______________________________________________ ChimeraX-users mailing list -- chimerax-users@cgl.ucsf.edu <mailto:chimerax-users@cgl.ucsf.edu> To unsubscribe send an email to chimerax-users-leave@cgl.ucsf.edu <mailto:chimerax-users-leave@cgl.ucsf.edu> Archives: https://mail.cgl.ucsf.edu/mailman/archives/list/chimerax-users@cgl.ucsf.edu/
_______________________________________________ ChimeraX-users mailing list -- chimerax-users@cgl.ucsf.edu <mailto:chimerax-users@cgl.ucsf.edu> To unsubscribe send an email to chimerax-users-leave@cgl.ucsf.edu <mailto:chimerax-users-leave@cgl.ucsf.edu> Archives: https://mail.cgl.ucsf.edu/mailman/archives/list/chimerax-users@cgl.ucsf.edu/
_______________________________________________ ChimeraX-users mailing list -- chimerax-users@cgl.ucsf.edu <mailto:chimerax-users@cgl.ucsf.edu> To unsubscribe send an email to chimerax-users-leave@cgl.ucsf.edu <mailto:chimerax-users-leave@cgl.ucsf.edu> Archives: https://mail.cgl.ucsf.edu/mailman/archives/list/chimerax-users@cgl.ucsf.edu/
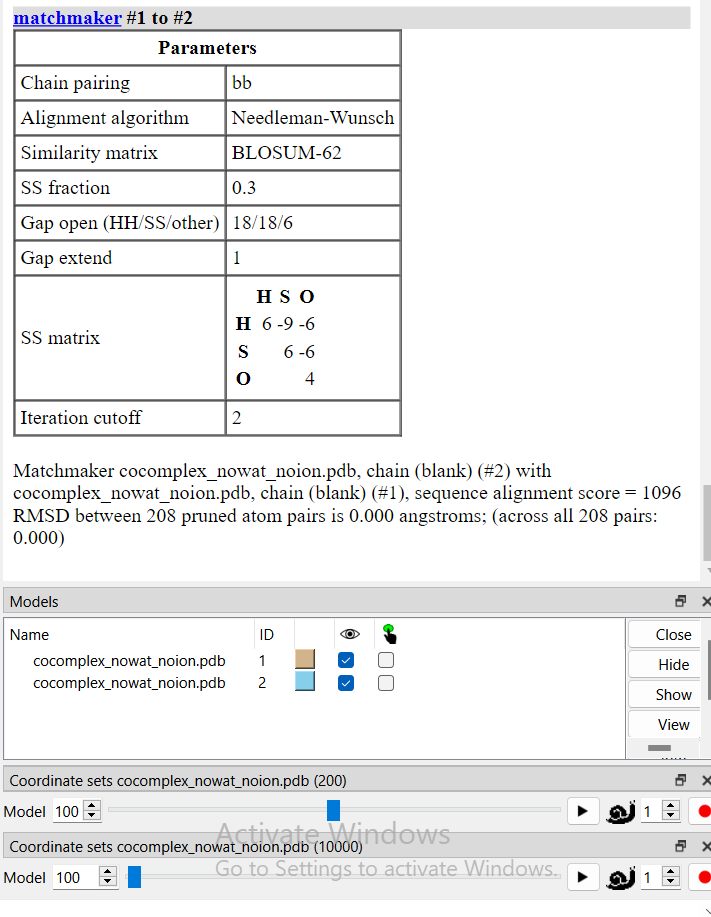
Thanks Eric, I opened them with these commands For Model #1 .: open "*\cocomplex_nowat_noion.pdb" ; open "*\md.nc" structureModel #1 step 50 For Model #2 .: open "*\cocomplex_nowat_noion.pdb" ; open "*\md.nc" structureModel #2 I've looked at the following frames Matching? Frames Step 50 Slider 200 frames #/200 frames Frames No extra options Slider 10k frames #/10k frames Yes 1 1 No 2 51 No 3 101 No 100 5001 No 200 9951 Matching? Frames Step 50 Slider 200 frames #/200 frames Frames No extra options Slider 10k frames #/10k frames Yes 1 1 Yes 2 2 Yes 3 3 Yes 100 100 Yes 200 200 I've also done matchmaker, and the RMSD between the 2 is 0 at the various frames of the second table. [cid:769c1c21-a5d6-4b3b-9ee4-b88d901cea27] Alex Lee Woods Lab ahl63010@uga.edu ________________________________ From: Eric Pettersen <pett@cgl.ucsf.edu> Sent: Monday, October 2, 2023 2:16 PM To: Alexander Hung Lee <Alexander.Lee@uga.edu> Cc: ChimeraX Users Help <chimerax-users@cgl.ucsf.edu> Subject: Re: [chimerax-users] Speeding up commands from cxc You don't often get email from pett@cgl.ucsf.edu. Learn why this is important<https://aka.ms/LearnAboutSenderIdentification> [EXTERNAL SENDER - PROCEED CAUTIOUSLY] Hi Alex, If I’m understanding the question correctly, no, frame 200 will not match frame 10,000. The first two commands you provided use the following frames from the overall trajectory: 1, 51, 101, … 9901, 9951, so the first frames will match, but the last frames will not. If you wanted the last frames to match then you would have to specify “start 50”, which case the 200 frames would be 50, 100, 150, … 9950, 10000. —Eric On Sep 30, 2023, at 7:19 PM, Alexander Hung Lee via ChimeraX-users <chimerax-users@cgl.ucsf.edu> wrote: Hi Eric, I've done the following commands on 3 .pdb files and used matchmaker on the 200th frame and 10,000th frame, respectively. open */md.nc structureModel #1 step 50 (slider shows 200 frames loaded) I've also tried open */md.nc structureModel #1 start 1 end 10000 step 50 (same result as above) vs open */md.nc structureModel #2 (slider shows 10,000 frames loaded - original) Both the 200/200 frames versions match the 200/10,000 frames version. If it worked, would 200/200 frames match 10,000/10,000 frames? Alex Lee Woods Lab ahl63010@uga.edu<mailto:ahl63010@uga.edu> ________________________________ From: Eric Pettersen <pett@cgl.ucsf.edu<mailto:pett@cgl.ucsf.edu>> Sent: Friday, September 29, 2023 4:54 PM To: Alexander Hung Lee <Alexander.Lee@uga.edu<mailto:Alexander.Lee@uga.edu>> Cc: ChimeraX Users Help <chimerax-users@cgl.ucsf.edu<mailto:chimerax-users@cgl.ucsf.edu>> Subject: Re: [chimerax-users] Speeding up commands from cxc You don't often get email from pett@cgl.ucsf.edu<mailto:pett@cgl.ucsf.edu>. Learn why this is important<https://aka.ms/LearnAboutSenderIdentification> [EXTERNAL SENDER - PROCEED CAUTIOUSLY] Hi Alex, I didn't even notice that you were already only showing every 50th frame. In that case you should use "step 50" in the open command and your play-through command becomes simply "coordset #1 1,200; wait 200". You have to realize that by default when you run the hbonds command on a trajectory model, it computes the H-bonds for every frame of the trajectory (controllable with the "coordsets" keyword). So using a trajectory of 200 frames vs. 10,000 will produce a 50x speedup, so the H-bonds calculation for 200 frames should take on the order of a half minute or so in your case. --Eric On Sep 28, 2023, at 8:18 PM, Alexander Hung Lee via ChimeraX-users <chimerax-users@cgl.ucsf.edu<mailto:chimerax-users@cgl.ucsf.edu>> wrote: Hi Eric, Thanks for the clarification. Little disappointed that hbond step can't leverage my hardware, but it's good to know I might be able to go around it by manipulating the frames. I'm using 10,000 frames for 100 ns MD simulation. Currently, my cxc workflow is producing ~ 1 min long movie by playing 10,000 frames in 50 frame increments and encoding at 4 fps. I'm visualizing the trajectory with "open *.pdb" followed by "open *.nc format Amber". If I were to use "open *.nc format Amber step 10" instead, would calculations be faster and I should change coordset #1 1,10000,50; wait 200 > coordset #1 1,1000,5; wait 200 ? Surface and hbond calculations should be significantly shorter (hbonds alone take 20 mins)? Thanks, Alex Lee Woods Lab ahl63010@uga.edu<mailto:ahl63010@uga.edu> ________________________________ From: Eric Pettersen <pett@cgl.ucsf.edu<mailto:pett@cgl.ucsf.edu>> Sent: Thursday, September 28, 2023 10:12 PM To: Alexander Hung Lee <Alexander.Lee@uga.edu<mailto:Alexander.Lee@uga.edu>> Cc: chimerax-users@cgl.ucsf.edu<mailto:chimerax-users@cgl.ucsf.edu> <chimerax-users@cgl.ucsf.edu<mailto:chimerax-users@cgl.ucsf.edu>> Subject: Re: [chimerax-users] Speeding up commands from cxc You don't often get email from pett@cgl.ucsf.edu<mailto:pett@cgl.ucsf.edu>. Learn why this is important<https://aka.ms/LearnAboutSenderIdentification> [EXTERNAL SENDER - PROCEED CAUTIOUSLY] Hi Alex, Yes, the bottlenecks are likely the surface recomputations as the trajectory plays, and the H-bonds calculation for 10,000 frames. I believe the surface calculation automatically uses multiple processors if available, so there's nothing you can do there, and the H-bonds calculation is very complicated and has not been parallelized (nor would it be easy to). My advice would be to use fewer frames. A 10,000 frame trajectory shown at 25 frames/sec would produce a movie close to 7 minutes long. Is that really what you want? If you used every 10th frame ("step 10" in the open command) then the movie would be 40 seconds long and take 3 minutes to record. --Eric Eric Pettersen UCSF Computer Graphics Lab On Sep 28, 2023, at 5:55 PM, Alexander Hung Lee via ChimeraX-users <chimerax-users@cgl.ucsf.edu<mailto:chimerax-users@cgl.ucsf.edu>> wrote: Hello everyone, I'm working to automate making movies from 10,000 frame trajectories. Currently, my cxc contains the following commands windowsize 1920 1080 set bgColor white view zalign ligand inFrontOf #1 sel protein surface; surface color salmon; transparency protein 60 target s sel :UNL color sel dark slate gray; color sel byhetero sel :SIA color sel dim gray; color sel byhetero sel :UNL hbonds :UNL reveal true surface zone #1 nearAtoms ligand distance 15 select zone ligand 15; select add ligand select ~sel ~cartoon sel delete sel ~sel view ligand movie record supersample 4 perframe "~label; label hbondatoms" ; coordset #1 1,10000,50; wait 200; ~perframe movie encode output hbond.mp4 bitrate 6000 framerate 4 The above takes ~ 30 mins, and it's something I would like to do for whole libraries I'm screening. I think the longest process is "hbonds :UNL reveal true" while second longest is movie record and perframe. Is there a way to speed this up? Is there a way to take advantage of multi-threading if it isn't already enabled? If multi-threading is already being used, what can I change to be more efficient? I deleted the parts that I didn't need "delete sel" since they're not involved in the binding site, and I thought it would help speed up surface calculations while recording. Regards, Alex Lee PhD Student Woods Research Group<https://woodsgroup.franklinresearch.uga.edu/> Department of Biochemistry & Molecular Biology Complex Carbohydrate Research Center University of Georgia 315 Riverbend Rd. Athens, GA 30602 Email: ahl63010@uga.edu<mailto:ahl63010@uga.edu> Mobile: (470) 641-3884 _______________________________________________ ChimeraX-users mailing list -- chimerax-users@cgl.ucsf.edu<mailto:chimerax-users@cgl.ucsf.edu> To unsubscribe send an email to chimerax-users-leave@cgl.ucsf.edu<mailto:chimerax-users-leave@cgl.ucsf.edu> Archives: https://mail.cgl.ucsf.edu/mailman/archives/list/chimerax-users@cgl.ucsf.edu/ _______________________________________________ ChimeraX-users mailing list -- chimerax-users@cgl.ucsf.edu<mailto:chimerax-users@cgl.ucsf.edu> To unsubscribe send an email to chimerax-users-leave@cgl.ucsf.edu<mailto:chimerax-users-leave@cgl.ucsf.edu> Archives: https://mail.cgl.ucsf.edu/mailman/archives/list/chimerax-users@cgl.ucsf.edu/ _______________________________________________ ChimeraX-users mailing list -- chimerax-users@cgl.ucsf.edu<mailto:chimerax-users@cgl.ucsf.edu> To unsubscribe send an email to chimerax-users-leave@cgl.ucsf.edu<mailto:chimerax-users-leave@cgl.ucsf.edu> Archives: https://mail.cgl.ucsf.edu/mailman/archives/list/chimerax-users@cgl.ucsf.edu/
{kind=link}
Hi Alex, Well, you are right — due to the way that the numpy Python library handles “slicing” into arrays, there was a bug in the code that produces the effect you got: the “step” results were just the beginning of the trajectory. I have committed a fix, so if you get tomorrow’s daily build (look for builds dated October 2 or later) then things should work as expected. —Eric
On Oct 2, 2023, at 11:54 AM, Alexander Hung Lee via ChimeraX-users <chimerax-users@cgl.ucsf.edu> wrote:
Thanks Eric,
I opened them with these commands For Model #1 .: open "*\cocomplex_nowat_noion.pdb" ; open "*\md.nc" structureModel #1 step 50 For Model #2 .: open "*\cocomplex_nowat_noion.pdb" ; open "*\md.nc" structureModel #2
I've looked at the following frames Matching? Frames Step 50 Slider 200 frames #/200 frames Frames No extra options Slider 10k frames #/10k frames Yes 1 1 No 2 51 No 3 101 No 100 5001 No 200 9951
Matching? Frames Step 50 Slider 200 frames #/200 frames Frames No extra options Slider 10k frames #/10k frames Yes 1 1 Yes 2 2 Yes 3 3 Yes 100 100 Yes 200 200
I've also done matchmaker, and the RMSD between the 2 is 0 at the various frames of the second table. <image.png>
Alex Lee Woods Lab ahl63010@uga.edu <mailto:ahl63010@uga.edu> From: Eric Pettersen <pett@cgl.ucsf.edu <mailto:pett@cgl.ucsf.edu>> Sent: Monday, October 2, 2023 2:16 PM To: Alexander Hung Lee <Alexander.Lee@uga.edu <mailto:Alexander.Lee@uga.edu>> Cc: ChimeraX Users Help <chimerax-users@cgl.ucsf.edu <mailto:chimerax-users@cgl.ucsf.edu>> Subject: Re: [chimerax-users] Speeding up commands from cxc
You don't often get email from pett@cgl.ucsf.edu <mailto:pett@cgl.ucsf.edu>. Learn why this is important <https://aka.ms/LearnAboutSenderIdentification> [EXTERNAL SENDER - PROCEED CAUTIOUSLY]
Hi Alex, If I’m understanding the question correctly, no, frame 200 will not match frame 10,000. The first two commands you provided use the following frames from the overall trajectory: 1, 51, 101, … 9901, 9951, so the first frames will match, but the last frames will not. If you wanted the last frames to match then you would have to specify “start 50”, which case the 200 frames would be 50, 100, 150, … 9950, 10000.
—Eric
On Sep 30, 2023, at 7:19 PM, Alexander Hung Lee via ChimeraX-users <chimerax-users@cgl.ucsf.edu> wrote:
Hi Eric,
I've done the following commands on 3 .pdb files and used matchmaker on the 200th frame and 10,000th frame, respectively.
open */md.nc structureModel #1 step 50 (slider shows 200 frames loaded) I've also tried open */md.nc structureModel #1 start 1 end 10000 step 50 (same result as above)
vs open */md.nc structureModel #2 (slider shows 10,000 frames loaded - original)
Both the 200/200 frames versions match the 200/10,000 frames version. If it worked, would 200/200 frames match 10,000/10,000 frames?
Alex Lee Woods Lab ahl63010@uga.edu <mailto:ahl63010@uga.edu>
From: Eric Pettersen <pett@cgl.ucsf.edu <mailto:pett@cgl.ucsf.edu>> Sent: Friday, September 29, 2023 4:54 PM To: Alexander Hung Lee <Alexander.Lee@uga.edu <mailto:Alexander.Lee@uga.edu>> Cc: ChimeraX Users Help <chimerax-users@cgl.ucsf.edu <mailto:chimerax-users@cgl.ucsf.edu>> Subject: Re: [chimerax-users] Speeding up commands from cxc
You don't often get email from pett@cgl.ucsf.edu <mailto:pett@cgl.ucsf.edu>. Learn why this is important <https://aka.ms/LearnAboutSenderIdentification> [EXTERNAL SENDER - PROCEED CAUTIOUSLY]
Hi Alex, I didn't even notice that you were already only showing every 50th frame. In that case you should use "step 50" in the open command and your play-through command becomes simply "coordset #1 1,200; wait 200". You have to realize that by default when you run the hbonds command on a trajectory model, it computes the H-bonds for every frame of the trajectory (controllable with the "coordsets" keyword). So using a trajectory of 200 frames vs. 10,000 will produce a 50x speedup, so the H-bonds calculation for 200 frames should take on the order of a half minute or so in your case.
--Eric
On Sep 28, 2023, at 8:18 PM, Alexander Hung Lee via ChimeraX-users <chimerax-users@cgl.ucsf.edu <mailto:chimerax-users@cgl.ucsf.edu>> wrote:
Hi Eric,
Thanks for the clarification. Little disappointed that hbond step can't leverage my hardware, but it's good to know I might be able to go around it by manipulating the frames. I'm using 10,000 frames for 100 ns MD simulation. Currently, my cxc workflow is producing ~ 1 min long movie by playing 10,000 frames in 50 frame increments and encoding at 4 fps.
I'm visualizing the trajectory with "open *.pdb" followed by "open *.nc format Amber". If I were to use "open *.nc format Amber step 10" instead, would calculations be faster and I should change coordset #1 1,10000,50; wait 200 > coordset #1 1,1000,5; wait 200 ? Surface and hbond calculations should be significantly shorter (hbonds alone take 20 mins)?
Thanks, Alex Lee Woods Lab ahl63010@uga.edu <mailto:ahl63010@uga.edu> From: Eric Pettersen <pett@cgl.ucsf.edu <mailto:pett@cgl.ucsf.edu>> Sent: Thursday, September 28, 2023 10:12 PM To: Alexander Hung Lee <Alexander.Lee@uga.edu <mailto:Alexander.Lee@uga.edu>> Cc: chimerax-users@cgl.ucsf.edu <mailto:chimerax-users@cgl.ucsf.edu> <chimerax-users@cgl.ucsf.edu <mailto:chimerax-users@cgl.ucsf.edu>> Subject: Re: [chimerax-users] Speeding up commands from cxc
You don't often get email from pett@cgl.ucsf.edu <mailto:pett@cgl.ucsf.edu>. Learn why this is important <https://aka.ms/LearnAboutSenderIdentification> [EXTERNAL SENDER - PROCEED CAUTIOUSLY]
Hi Alex, Yes, the bottlenecks are likely the surface recomputations as the trajectory plays, and the H-bonds calculation for 10,000 frames. I believe the surface calculation automatically uses multiple processors if available, so there's nothing you can do there, and the H-bonds calculation is very complicated and has not been parallelized (nor would it be easy to). My advice would be to use fewer frames. A 10,000 frame trajectory shown at 25 frames/sec would produce a movie close to 7 minutes long. Is that really what you want? If you used every 10th frame ("step 10" in the open command) then the movie would be 40 seconds long and take 3 minutes to record.
--Eric
Eric Pettersen UCSF Computer Graphics Lab
On Sep 28, 2023, at 5:55 PM, Alexander Hung Lee via ChimeraX-users <chimerax-users@cgl.ucsf.edu <mailto:chimerax-users@cgl.ucsf.edu>> wrote:
Hello everyone,
I'm working to automate making movies from 10,000 frame trajectories. Currently, my cxc contains the following commands
windowsize 1920 1080 set bgColor white view zalign ligand inFrontOf #1 sel protein surface; surface color salmon; transparency protein 60 target s sel :UNL color sel dark slate gray; color sel byhetero sel :SIA color sel dim gray; color sel byhetero sel :UNL hbonds :UNL reveal true surface zone #1 nearAtoms ligand distance 15 select zone ligand 15; select add ligand select ~sel ~cartoon sel delete sel ~sel view ligand movie record supersample 4 perframe "~label; label hbondatoms" ; coordset #1 1,10000,50; wait 200; ~perframe movie encode output hbond.mp4 bitrate 6000 framerate 4
The above takes ~ 30 mins, and it's something I would like to do for whole libraries I'm screening. I think the longest process is "hbonds :UNL reveal true" while second longest is movie record and perframe. Is there a way to speed this up? Is there a way to take advantage of multi-threading if it isn't already enabled? If multi-threading is already being used, what can I change to be more efficient?
I deleted the parts that I didn't need "delete sel" since they're not involved in the binding site, and I thought it would help speed up surface calculations while recording.
Regards, Alex Lee PhD Student Woods Research Group <https://woodsgroup.franklinresearch.uga.edu/> Department of Biochemistry & Molecular Biology Complex Carbohydrate Research Center University of Georgia
315 Riverbend Rd. Athens, GA 30602 Email: ahl63010@uga.edu <mailto:ahl63010@uga.edu> Mobile: (470) 641-3884 _______________________________________________ ChimeraX-users mailing list -- chimerax-users@cgl.ucsf.edu <mailto:chimerax-users@cgl.ucsf.edu> To unsubscribe send an email to chimerax-users-leave@cgl.ucsf.edu <mailto:chimerax-users-leave@cgl.ucsf.edu> Archives: https://mail.cgl.ucsf.edu/mailman/archives/list/chimerax-users@cgl.ucsf.edu/
_______________________________________________ ChimeraX-users mailing list -- chimerax-users@cgl.ucsf.edu <mailto:chimerax-users@cgl.ucsf.edu> To unsubscribe send an email to chimerax-users-leave@cgl.ucsf.edu <mailto:chimerax-users-leave@cgl.ucsf.edu> Archives: https://mail.cgl.ucsf.edu/mailman/archives/list/chimerax-users@cgl.ucsf.edu/
_______________________________________________ ChimeraX-users mailing list -- chimerax-users@cgl.ucsf.edu <mailto:chimerax-users@cgl.ucsf.edu> To unsubscribe send an email to chimerax-users-leave@cgl.ucsf.edu <mailto:chimerax-users-leave@cgl.ucsf.edu> Archives: https://mail.cgl.ucsf.edu/mailman/archives/list/chimerax-users@cgl.ucsf.edu/
_______________________________________________ ChimeraX-users mailing list -- chimerax-users@cgl.ucsf.edu <mailto:chimerax-users@cgl.ucsf.edu> To unsubscribe send an email to chimerax-users-leave@cgl.ucsf.edu <mailto:chimerax-users-leave@cgl.ucsf.edu> Archives: https://mail.cgl.ucsf.edu/mailman/archives/list/chimerax-users@cgl.ucsf.edu/
participants (2)
-
 Alexander Hung Lee
Alexander Hung Lee -
 Eric Pettersen
Eric Pettersen