Advice on handling cofactor-protein electrostatic interactions

Hi, I would like to present effects of mutations around cofactors (TDP derivatives) on the protein - cofactors electrostatic interactions. I use APBS and chimerax to display the electrostatic surface potential of the protein, but since PDB2PQR eliminates the cofactor from the pqr file I need a tool for the cofactor surface potential. I'm wondering what tool I should use for this. Is it Antechamber+chimerax or something else? Is there by chance a demo for such scenario? I'd appreciate your advice. Cheers, Boaz Boaz Shaanan, Ph.D. Dept. of Life Sciences Ben-Gurion University of the Negev Beer-Sheva 84105 Israel E-mail: bshaanan@bgu.ac.il Phone: 972-8-647-2220 Fax: 972-8-647-2992 or 972-8-646-1710

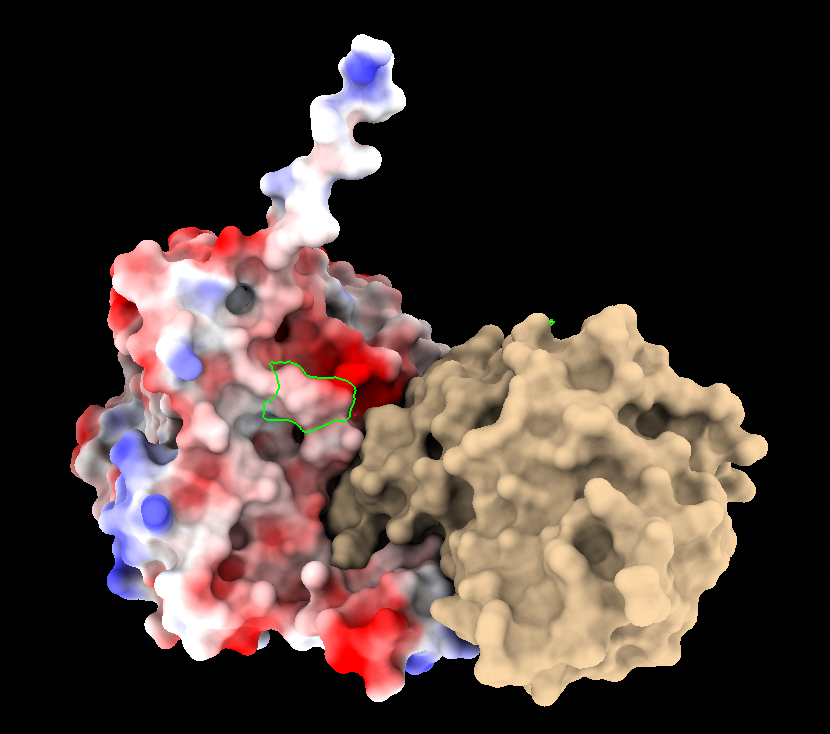
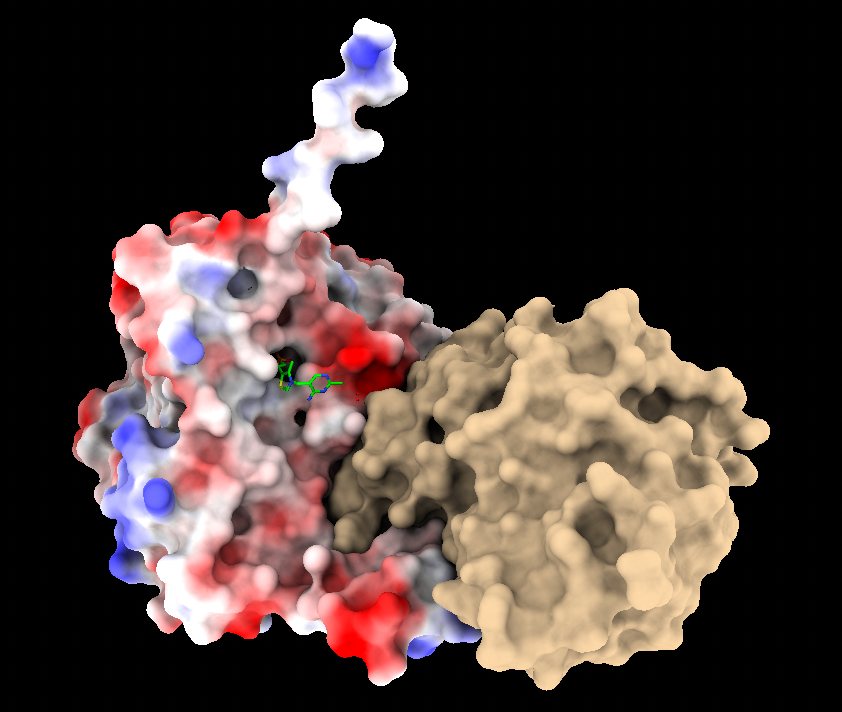
Hi Boaz, This answer addresses coloring by electrostatic potential (ESP). Calculating the electrostatic interaction per se (energy?) is somewhat different and not something that ChimeraX does. ChimeraX can color some surface, which could be a molecular surface, by the values in a 3D map, which could be electrostatic potential (ESP). See "color electrostatic" <https://rbvi.ucsf.edu/chimerax/docs/user/commands/color.html#map> The ESP map can come from a Poisson-Boltzmann calculation (APBS, DelPhi, others) or from a more simplistic Coulombic calculation which can be done directly in ChimeraX. Poisson-Boltzmann calculations require atomic coordinates, partial charges, and radii, but the different programs differ in how you input these values. If I recall correctly, APBS takes as input a pqr file (q means charge, r means radii) which would usually be generated by PDB2PQR, but you said PDB2PQR throws out the nonstandard residues like cofactors, I guess because it doesn't have charges for them. ChimeraX assigns radii and can assign atomic partial charges (via lookup table for standard residues and Antechamber calculation for nonstandard residues like cofactors). However, ChimeraX does not write pqr format. Also in ChimeraX, these partial atomic charges are for all atoms (i.e. all hydrogens explicit) and not a united-atom model (in which hydrogens on carbons are merged with those carbons so that they become implicit) which is often used for Poisson-Boltzmann calculations. So if you wanted to go the APBS route, in ChimeraX you would add hydrogens and charges, then save the cofactor radii and atomic partial charges (atom attributes "radius" and "charge") in the attribute file format, and if you were using a united-atom charge model, manually generate united atoms from CH, CH2, and CH3 groups in the cofactor by adding the hydrogen charges to those of the carbons where they are attached and figuring out the appropriate united-atom radii, then reformat the cofactor atom coordinates, radii and charge values into pqr format and then add these lines to your protein pqr file from PDB2PQR, then use the resulting combined pqr file as input to APBS to get the ESP map that includes cofactor contributions. Actually I don't even know if APBS would accept that pqr file or not, but that is what I imagine you would try. So let's assume the APBS calculation will run using that pqr file. Then you would open the APBS ESP map that includes cofactor contributions in ChimeraX and color the molecular surface. If the cofactor is covalently attached to the protein then the protein molecular surface may already include it. However, if the cofactor is not covalently attached, you would need to tell ChimeraX specifically to lump it inside of the same surface as the protein with the "include" option of "surface" when you are calculating and displaying the molecular surface. The "coulombic" approach wil do more of these steps for you, given the "include" trick as well as the "surfaces" option of the "coulombic" command, but calculating ESP with the relatively simplistic Coulomb's law method. For example, 1qs0 chain A includes a noncovalently bound TPP residue and I could show the surface including the TPP and colored including its potential contribution: open 1qs0 surf include :tpp coulombic surfaces #1.2 <https://rbvi.ucsf.edu/chimerax/docs/user/commands/surface.html#options> <https://rbvi.ucsf.edu/chimerax/docs/user/commands/coulombic.html> See first image attached with :tpp selected (green outline surface patch) If you don't use the "include" and "surfaces" options in these commands, the surface will be redrawn to include only the protein part with the TPP outside of it, like other ligands and the Coulombic ESP will not include the TPP contribution. See second image attached. I hope this helps, Elaine ----- Elaine C. Meng, Ph.D. UCSF Chimera(X) team Department of Pharmaceutical Chemistry University of California, San Francisco
On Aug 7, 2023, at 4:10 PM, Boaz Shaanan via ChimeraX-users <chimerax-users@cgl.ucsf.edu> wrote:
Hi,
I would like to present effects of mutations around cofactors (TDP derivatives) on the protein - cofactors electrostatic interactions. I use APBS and chimerax to display the electrostatic surface potential of the protein, but since PDB2PQR eliminates the cofactor from the pqr file I need a tool for the cofactor surface potential. I'm wondering what tool I should use for this. Is it Antechamber+chimerax or something else? Is there by chance a demo for such scenario? I'd appreciate your advice. Cheers,
Boaz
Boaz Shaanan, Ph.D. Dept. of Life Sciences Ben-Gurion University of the Negev Beer-Sheva 84105 Israel
E-mail: bshaanan@bgu.ac.il Phone: 972-8-647-2220 Fax: 972-8-647-2992 or 972-8-646-1710
_______________________________________________ ChimeraX-users mailing list -- chimerax-users@cgl.ucsf.edu To unsubscribe send an email to chimerax-users-leave@cgl.ucsf.edu Archives: https://mail.cgl.ucsf.edu/mailman/archives/list/chimerax-users@cgl.ucsf.edu/
{kind=link}
{kind=link}
Hi Elaine, Thanks a lot for the detailed reply and (as always) very helpful suggestions. I'll experiment first with the Chimerax coulombic surfaces and trick to include the cofactor. This sounds attractive, certainly for the TDP case as a starting point. As for derivatives of TDP, which are already included in the pdb, I guess I should follow the 'Add Charges' guide, right? Thanks again. Boaz Boaz Shaanan, Ph.D. Dept. of Life Sciences Ben-Gurion University of the Negev Beer-Sheva 84105 Israel E-mail: bshaanan@bgu.ac.il Phone: 972-8-647-2220 Fax: 972-8-647-2992 or 972-8-646-1710 ________________________________ From: Elaine Meng <meng@cgl.ucsf.edu> Sent: Monday, August 7, 2023 10:17 PM To: ��� ���� <bshaanan@bgu.ac.il> Cc: chimerax-users@cgl.ucsf.edu <chimerax-users@cgl.ucsf.edu> Subject: Re: [chimerax-users] Advice on handling cofactor-protein electrostatic interactions Hi Boaz, This answer addresses coloring by electrostatic potential (ESP). Calculating the electrostatic interaction per se (energy?) is somewhat different and not something that ChimeraX does. ChimeraX can color some surface, which could be a molecular surface, by the values in a 3D map, which could be electrostatic potential (ESP). See "color electrostatic" <https://rbvi.ucsf.edu/chimerax/docs/user/commands/color.html#map<https://imsva91-ctp.trendmicro.com:443/wis/clicktime/v1/query?url=https%3a%2f%2frbvi.ucsf.edu%2fchimerax%2fdocs%2fuser%2fcommands%2fcolor.html%23map&umid=8D1517BE-025F-F806-8BB4-612BD1B2C2E9&auth=701951cf8dfb7cd99762e0f5be252082d875f1cf-9c202d037c23b2a6ecd19737a2ab89370faadf8c>> The ESP map can come from a Poisson-Boltzmann calculation (APBS, DelPhi, others) or from a more simplistic Coulombic calculation which can be done directly in ChimeraX. Poisson-Boltzmann calculations require atomic coordinates, partial charges, and radii, but the different programs differ in how you input these values. If I recall correctly, APBS takes as input a pqr file (q means charge, r means radii) which would usually be generated by PDB2PQR, but you said PDB2PQR throws out the nonstandard residues like cofactors, I guess because it doesn't have charges for them. ChimeraX assigns radii and can assign atomic partial charges (via lookup table for standard residues and Antechamber calculation for nonstandard residues like cofactors). However, ChimeraX does not write pqr format. Also in ChimeraX, these partial atomic charges are for all atoms (i.e. all hydrogens explicit) and not a united-atom model (in which hydrogens on carbons are merged with those carbons so that they become implicit) which is often used for Poisson-Boltzmann calculations. So if you wanted to go the APBS route, in ChimeraX you would add hydrogens and charges, then save the cofactor radii and atomic partial charges (atom attributes "radius" and "charge") in the attribute file format, and if you were using a united-atom charge model, manually generate united atoms from CH, CH2, and CH3 groups in the cofactor by adding the hydrogen charges to those of the carbons where they are attached and figuring out the appropriate united-atom radii, then reformat the cofactor atom coordinates, radii and charge values into pqr format and then add these lines to your protein pqr file from PDB2PQR, then use the resulting combined pqr file as input to APBS to get the ESP map that includes cofactor contributions. Actually I don't even know if APBS would accept that pqr file or not, but that is what I imagine you would try. So let's assume the APBS calculation will run using that pqr file. Then you would open the APBS ESP map that includes cofactor contributions in ChimeraX and color the molecular surface. If the cofactor is covalently attached to the protein then the protein molecular surface may already include it. However, if the cofactor is not covalently attached, you would need to tell ChimeraX specifically to lump it inside of the same surface as the protein with the "include" option of "surface" when you are calculating and displaying the molecular surface. The "coulombic" approach wil do more of these steps for you, given the "include" trick as well as the "surfaces" option of the "coulombic" command, but calculating ESP with the relatively simplistic Coulomb's law method. For example, 1qs0 chain A includes a noncovalently bound TPP residue and I could show the surface including the TPP and colored including its potential contribution: open 1qs0 surf include :tpp coulombic surfaces #1.2 <https://rbvi.ucsf.edu/chimerax/docs/user/commands/surface.html#options<https://imsva91-ctp.trendmicro.com:443/wis/clicktime/v1/query?url=https%3a%2f%2frbvi.ucsf.edu%2fchimerax%2fdocs%2fuser%2fcommands%2fsurface.html%23options&umid=8D1517BE-025F-F806-8BB4-612BD1B2C2E9&auth=701951cf8dfb7cd99762e0f5be252082d875f1cf-797a08f02e62255329eb3e1a59116084790684c3>> <https://rbvi.ucsf.edu/chimerax/docs/user/commands/coulombic.html<https://imsva91-ctp.trendmicro.com:443/wis/clicktime/v1/query?url=https%3a%2f%2frbvi.ucsf.edu%2fchimerax%2fdocs%2fuser%2fcommands%2fcoulombic.html&umid=8D1517BE-025F-F806-8BB4-612BD1B2C2E9&auth=701951cf8dfb7cd99762e0f5be252082d875f1cf-9ddb0b7ca46a5d4a65498ab0338d10b8b8657f0f>> See first image attached with :tpp selected (green outline surface patch) If you don't use the "include" and "surfaces" options in these commands, the surface will be redrawn to include only the protein part with the TPP outside of it, like other ligands and the Coulombic ESP will not include the TPP contribution. See second image attached. I hope this helps, Elaine ----- Elaine C. Meng, Ph.D. UCSF Chimera(X) team Department of Pharmaceutical Chemistry University of California, San Francisco [cid:786869E3-E870-47BD-8806-48E579AD5139][cid:02D440C4-E382-43E6-8A4B-EF20EEAAA7CF] On Aug 7, 2023, at 4:10 PM, Boaz Shaanan via ChimeraX-users <chimerax-users@cgl.ucsf.edu<mailto:chimerax-users@cgl.ucsf.edu>> wrote: Hi, I would like to present effects of mutations around cofactors (TDP derivatives) on the protein - cofactors electrostatic interactions. I use APBS and chimerax to display the electrostatic surface potential of the protein, but since PDB2PQR eliminates the cofactor from the pqr file I need a tool for the cofactor surface potential. I'm wondering what tool I should use for this. Is it Antechamber+chimerax or something else? Is there by chance a demo for such scenario? I'd appreciate your advice. Cheers, Boaz Boaz Shaanan, Ph.D. Dept. of Life Sciences Ben-Gurion University of the Negev Beer-Sheva 84105 Israel E-mail: bshaanan@bgu.ac.il<mailto:bshaanan@bgu.ac.il> Phone: 972-8-647-2220 Fax: 972-8-647-2992 or 972-8-646-1710 _______________________________________________ ChimeraX-users mailing list -- chimerax-users@cgl.ucsf.edu To unsubscribe send an email to chimerax-users-leave@cgl.ucsf.edu Archives: https://imsva91-ctp.trendmicro.com:443/wis/clicktime/v1/query?url=https%3a%2...
Hi Boaz, (apologies if you get this multiple times, the chimerax-users list was rejecting my earlier replies for some unknown reason) If you use Coulombic it will automatically calculate the charges, as in my example of 1qs0 with the TPP residue. I.e. you don't have to do it in a separate step first, and in fact it may be better if you do not: if you use Add Charge first, it will also add hydrogens and make the surface a lot bumpier. If you just use Coulombic the hydrogens are added only on a temporary copy that is in memory, not on your model, and the coloring will be applied to the smoother molecular surface without hydrogens. You just need to substitute the cofactor residue name for ":tpp" in my example, if it is not TPP. I hope this helps, Elaine ----- Elaine C. Meng, Ph.D. UCSF Chimera(X) team Department of Pharmaceutical Chemistry University of California, San Francisco
On Aug 8, 2023, at 7:09 AM, Boaz Shaanan via ChimeraX-users <chimerax-users@cgl.ucsf.edu> wrote:
Hi Elaine,
Thanks a lot for the detailed reply and (as always) very helpful suggestions. I'll experiment first with the Chimerax coulombic surfaces and trick to include the cofactor. This sounds attractive, certainly for the TDP case as a starting point. As for derivatives of TDP, which are already included in the pdb, I guess I should follow the 'Add Charges' guide, right?
Thanks again.
Boaz
Aha, Last night it happened to me too (in fact I saw 3 copies of my message to you/list). Strange. Boaz Boaz Shaanan, Ph.D. Dept. of Life Sciences Ben-Gurion University of the Negev Beer-Sheva 84105 Israel E-mail: bshaanan@bgu.ac.il Phone: 972-8-647-2220 Fax: 972-8-647-2992 or 972-8-646-1710 ________________________________ From: Elaine Meng <meng@cgl.ucsf.edu> Sent: Tuesday, August 8, 2023 12:22 PM To: ��� ���� <bshaanan@bgu.ac.il> Cc: ChimeraX Users Help <chimerax-users@cgl.ucsf.edu> Subject: Re: [chimerax-users] Advice on handling cofactor-protein electrostatic interactions Hi Boaz, (apologies if you get this multiple times, the chimerax-users list was rejecting my earlier replies for some unknown reason) If you use Coulombic it will automatically calculate the charges, as in my example of 1qs0 with the TPP residue. I.e. you don't have to do it in a separate step first, and in fact it may be better if you do not: if you use Add Charge first, it will also add hydrogens and make the surface a lot bumpier. If you just use Coulombic the hydrogens are added only on a temporary copy that is in memory, not on your model, and the coloring will be applied to the smoother molecular surface without hydrogens. You just need to substitute the cofactor residue name for ":tpp" in my example, if it is not TPP. I hope this helps, Elaine ----- Elaine C. Meng, Ph.D. UCSF Chimera(X) team Department of Pharmaceutical Chemistry University of California, San Francisco
On Aug 8, 2023, at 7:09 AM, Boaz Shaanan via ChimeraX-users <chimerax-users@cgl.ucsf.edu> wrote:
Hi Elaine,
Thanks a lot for the detailed reply and (as always) very helpful suggestions. I'll experiment first with the Chimerax coulombic surfaces and trick to include the cofactor. This sounds attractive, certainly for the TDP case as a starting point. As for derivatives of TDP, which are already included in the pdb, I guess I should follow the 'Add Charges' guide, right?
Thanks again.
Boaz
Hi Elaine, Your advice on using the Coulomb surface +vcofactor works fine, thanks. I have a few more questions though: 1. I'm trying to figure out the difference between the Coulomb surface with and w/o the coafactor but I'm not sure I see a difference. Is there an option to 'walk' with the mouse on the surface to probe the actual values (as in the surface generates by the volume viewer)? 2. Writing out the displayed model coordinates in mol2 format does not give an option write out a selected displayed group (e.g. cofactors) as in the case of .pdb. I guess this has to do with mol2 format but is it not possible to add such an option? Thanks, Boaz Boaz Shaanan, Ph.D. Dept. of Life Sciences Ben-Gurion University of the Negev Beer-Sheva 84105 Israel E-mail: bshaanan@bgu.ac.il Phone: 972-8-647-2220 Fax: 972-8-647-2992 or 972-8-646-1710 ________________________________ From: ��� ���� <bshaanan@bgu.ac.il> Sent: Tuesday, August 8, 2023 12:25 PM To: ChimeraX Users Help <chimerax-users@cgl.ucsf.edu> Subject: Re: [chimerax-users] Advice on handling cofactor-protein electrostatic interactions Aha, Last night it happened to me too (in fact I saw 3 copies of my message to you/list). Strange. Boaz Boaz Shaanan, Ph.D. Dept. of Life Sciences Ben-Gurion University of the Negev Beer-Sheva 84105 Israel E-mail: bshaanan@bgu.ac.il Phone: 972-8-647-2220 Fax: 972-8-647-2992 or 972-8-646-1710 ________________________________ From: Elaine Meng <meng@cgl.ucsf.edu> Sent: Tuesday, August 8, 2023 12:22 PM To: ��� ���� <bshaanan@bgu.ac.il> Cc: ChimeraX Users Help <chimerax-users@cgl.ucsf.edu> Subject: Re: [chimerax-users] Advice on handling cofactor-protein electrostatic interactions Hi Boaz, (apologies if you get this multiple times, the chimerax-users list was rejecting my earlier replies for some unknown reason) If you use Coulombic it will automatically calculate the charges, as in my example of 1qs0 with the TPP residue. I.e. you don't have to do it in a separate step first, and in fact it may be better if you do not: if you use Add Charge first, it will also add hydrogens and make the surface a lot bumpier. If you just use Coulombic the hydrogens are added only on a temporary copy that is in memory, not on your model, and the coloring will be applied to the smoother molecular surface without hydrogens. You just need to substitute the cofactor residue name for ":tpp" in my example, if it is not TPP. I hope this helps, Elaine ----- Elaine C. Meng, Ph.D. UCSF Chimera(X) team Department of Pharmaceutical Chemistry University of California, San Francisco
On Aug 8, 2023, at 7:09 AM, Boaz Shaanan via ChimeraX-users <chimerax-users@cgl.ucsf.edu> wrote:
Hi Elaine,
Thanks a lot for the detailed reply and (as always) very helpful suggestions. I'll experiment first with the Chimerax coulombic surfaces and trick to include the cofactor. This sounds attractive, certainly for the TDP case as a starting point. As for derivatives of TDP, which are already included in the pdb, I guess I should follow the 'Add Charges' guide, right?
Thanks again.
Boaz
Hi Boaz, (1) I thought we'd answered this before and actually, you were the one who asked! See this previous reply about setting an option in Surface Color to show value in a pop-up on mouseover <https://mail.cgl.ucsf.edu/mailman/archives/list/chimerax-users@cgl.ucsf.edu/...> Of course, the shape of the surface will also be different between when the cofactor is on the inside of it and on the outside of it. (2) as long as you have a saved copy of the entire thing, just delete the part you don't want before saving. I hope this helps, Elaine ----- Elaine C. Meng, Ph.D. UCSF Chimera(X) team Department of Pharmaceutical Chemistry University of California, San Francisco
On Sep 12, 2023, at 9:25 AM, Boaz Shaanan via ChimeraX-users <chimerax-users@cgl.ucsf.edu> wrote:
Hi Elaine,
Your advice on using the Coulomb surface +vcofactor works fine, thanks. I have a few more questions though: • I'm trying to figure out the difference between the Coulomb surface with and w/o the coafactor but I'm not sure I see a difference. Is there an option to 'walk' with the mouse on the surface to probe the actual values (as in the surface generates by the volume viewer)? • Writing out the displayed model coordinates in mol2 format does not give an option write out a selected displayed group (e.g. cofactors) as in the case of .pdb. I guess this has to do with mol2 format but is it not possible to add such an option?
Thanks,
Boaz Boaz Shaanan, Ph.D. Dept. of Life Sciences Ben-Gurion University of the Negev Beer-Sheva 84105 Israel
E-mail: bshaanan@bgu.ac.il Phone: 972-8-647-2220 Fax: 972-8-647-2992 or 972-8-646-1710
From: בעז שאנן <bshaanan@bgu.ac.il> Sent: Tuesday, August 8, 2023 12:25 PM To: ChimeraX Users Help <chimerax-users@cgl.ucsf.edu> Subject: Re: [chimerax-users] Advice on handling cofactor-protein electrostatic interactions
Aha, Last night it happened to me too (in fact I saw 3 copies of my message to you/list). Strange. Boaz
Boaz Shaanan, Ph.D. Dept. of Life Sciences Ben-Gurion University of the Negev Beer-Sheva 84105 Israel
E-mail: bshaanan@bgu.ac.il Phone: 972-8-647-2220 Fax: 972-8-647-2992 or 972-8-646-1710
From: Elaine Meng <meng@cgl.ucsf.edu> Sent: Tuesday, August 8, 2023 12:22 PM To: בעז שאנן <bshaanan@bgu.ac.il> Cc: ChimeraX Users Help <chimerax-users@cgl.ucsf.edu> Subject: Re: [chimerax-users] Advice on handling cofactor-protein electrostatic interactions
Hi Boaz, (apologies if you get this multiple times, the chimerax-users list was rejecting my earlier replies for some unknown reason)
If you use Coulombic it will automatically calculate the charges, as in my example of 1qs0 with the TPP residue. I.e. you don't have to do it in a separate step first, and in fact it may be better if you do not: if you use Add Charge first, it will also add hydrogens and make the surface a lot bumpier. If you just use Coulombic the hydrogens are added only on a temporary copy that is in memory, not on your model, and the coloring will be applied to the smoother molecular surface without hydrogens.
You just need to substitute the cofactor residue name for ":tpp" in my example, if it is not TPP. I hope this helps, Elaine ----- Elaine C. Meng, Ph.D. UCSF Chimera(X) team Department of Pharmaceutical Chemistry University of California, San Francisco
On Aug 8, 2023, at 7:09 AM, Boaz Shaanan via ChimeraX-users <chimerax-users@cgl.ucsf.edu> wrote:
Hi Elaine,
Thanks a lot for the detailed reply and (as always) very helpful suggestions. I'll experiment first with the Chimerax coulombic surfaces and trick to include the cofactor. This sounds attractive, certainly for the TDP case as a starting point. As for derivatives of TDP, which are already included in the pdb, I guess I should follow the 'Add Charges' guide, right?
Thanks again.
Boaz
_______________________________________________ ChimeraX-users mailing list -- chimerax-users@cgl.ucsf.edu To unsubscribe send an email to chimerax-users-leave@cgl.ucsf.edu Archives: https://mail.cgl.ucsf.edu/mailman/archives/list/chimerax-users@cgl.ucsf.edu/
Hi Boaz, Addendum to (2). Actually there are options to the "save" command with which you can do this without having to delete anything beforehand. See the "atoms" and/or "skipAtoms" options: <https://rbvi.ucsf.edu/chimerax/docs/user/commands/save.html#mol2> Maybe you were looking at the File... Save dialog, which does not currently expose those options when the Mol2 file type is chosen. Best, Elaine
On Sep 12, 2023, at 11:10 AM, Elaine Meng via ChimeraX-users <chimerax-users@cgl.ucsf.edu> wrote:
Hi Boaz, (1) I thought we'd answered this before and actually, you were the one who asked! See this previous reply about setting an option in Surface Color to show value in a pop-up on mouseover
<https://mail.cgl.ucsf.edu/mailman/archives/list/chimerax-users@cgl.ucsf.edu/...>
Of course, the shape of the surface will also be different between when the cofactor is on the inside of it and on the outside of it.
(2) as long as you have a saved copy of the entire thing, just delete the part you don't want before saving.
I hope this helps, Elaine ----- Elaine C. Meng, Ph.D. UCSF Chimera(X) team Department of Pharmaceutical Chemistry University of California, San Francisco
On Sep 12, 2023, at 9:25 AM, Boaz Shaanan via ChimeraX-users <chimerax-users@cgl.ucsf.edu> wrote:
Hi Elaine,
Your advice on using the Coulomb surface +vcofactor works fine, thanks. I have a few more questions though: • I'm trying to figure out the difference between the Coulomb surface with and w/o the coafactor but I'm not sure I see a difference. Is there an option to 'walk' with the mouse on the surface to probe the actual values (as in the surface generates by the volume viewer)? • Writing out the displayed model coordinates in mol2 format does not give an option write out a selected displayed group (e.g. cofactors) as in the case of .pdb. I guess this has to do with mol2 format but is it not possible to add such an option?
Thanks,
Boaz Boaz Shaanan, Ph.D. Dept. of Life Sciences Ben-Gurion University of the Negev Beer-Sheva 84105 Israel
E-mail: bshaanan@bgu.ac.il Phone: 972-8-647-2220 Fax: 972-8-647-2992 or 972-8-646-1710
From: בעז שאנן <bshaanan@bgu.ac.il> Sent: Tuesday, August 8, 2023 12:25 PM To: ChimeraX Users Help <chimerax-users@cgl.ucsf.edu> Subject: Re: [chimerax-users] Advice on handling cofactor-protein electrostatic interactions
Aha, Last night it happened to me too (in fact I saw 3 copies of my message to you/list). Strange. Boaz
Boaz Shaanan, Ph.D. Dept. of Life Sciences Ben-Gurion University of the Negev Beer-Sheva 84105 Israel
E-mail: bshaanan@bgu.ac.il Phone: 972-8-647-2220 Fax: 972-8-647-2992 or 972-8-646-1710
From: Elaine Meng <meng@cgl.ucsf.edu> Sent: Tuesday, August 8, 2023 12:22 PM To: בעז שאנן <bshaanan@bgu.ac.il> Cc: ChimeraX Users Help <chimerax-users@cgl.ucsf.edu> Subject: Re: [chimerax-users] Advice on handling cofactor-protein electrostatic interactions
Hi Boaz, (apologies if you get this multiple times, the chimerax-users list was rejecting my earlier replies for some unknown reason)
If you use Coulombic it will automatically calculate the charges, as in my example of 1qs0 with the TPP residue. I.e. you don't have to do it in a separate step first, and in fact it may be better if you do not: if you use Add Charge first, it will also add hydrogens and make the surface a lot bumpier. If you just use Coulombic the hydrogens are added only on a temporary copy that is in memory, not on your model, and the coloring will be applied to the smoother molecular surface without hydrogens.
You just need to substitute the cofactor residue name for ":tpp" in my example, if it is not TPP. I hope this helps, Elaine ----- Elaine C. Meng, Ph.D. UCSF Chimera(X) team Department of Pharmaceutical Chemistry University of California, San Francisco
On Aug 8, 2023, at 7:09 AM, Boaz Shaanan via ChimeraX-users <chimerax-users@cgl.ucsf.edu> wrote:
Hi Elaine,
Thanks a lot for the detailed reply and (as always) very helpful suggestions. I'll experiment first with the Chimerax coulombic surfaces and trick to include the cofactor. This sounds attractive, certainly for the TDP case as a starting point. As for derivatives of TDP, which are already included in the pdb, I guess I should follow the 'Add Charges' guide, right?
Thanks again.
Boaz
_______________________________________________ ChimeraX-users mailing list -- chimerax-users@cgl.ucsf.edu To unsubscribe send an email to chimerax-users-leave@cgl.ucsf.edu Archives: https://mail.cgl.ucsf.edu/mailman/archives/list/chimerax-users@cgl.ucsf.edu/
_______________________________________________ ChimeraX-users mailing list -- chimerax-users@cgl.ucsf.edu To unsubscribe send an email to chimerax-users-leave@cgl.ucsf.edu Archives: https://mail.cgl.ucsf.edu/mailman/archives/list/chimerax-users@cgl.ucsf.edu/
participants (2)
-
 Boaz Shaanan
Boaz Shaanan -
 Elaine Meng
Elaine Meng