
Hi, The intracellular domain (ICD) of Alpha7 nicotinic receptors, like almost all eukaryotic Cys-loop receptors, is not resolved using X-ray crystallography or cryo-EM. Last week the PDB for an ensemble of NMR ICD structures for the receptor was released (PDB 7RPM). I've been unsuccessfully trying to use ChimeraX and Modeller to make a model of a subunit that includes the ICD. The 7RPM models include all 4 transmembrane domains and the ICD (but lacks the extracellular domain), so there is considerable overlap with the 7KOO PDB fo a7. I started by deleting all but one of the identical subunits in the 7KOO pdb and loaded one of the 7RPM models and tried to use Modeller to make a construct that includes both the extracellular domain and the ICD. Unfortunately, even though both 7KOO and the single 7RPM are in the sequence alignments window, the ChimeraX-Modeller combination ignores the structure that is not chosen as the target (e.g. if 7RPM is the target, there is no extracellular domain, but if 7KOO is the target, there is either no ICD or the ICD that is shown bears no relevance to the structure in 7RPM). I can align the two structures well with a low RMSD using Matchmaker because of the TMDs overlapping. I just want to fill in the missing parts. Is there a special trick to get Modeller to fill in the missing parts using ChimeraX as the interface? I'm not that experienced using Modeller. Thanks, Ralph H. Loring Associate Professor of Pharmacology Department of Pharmaceutical Sciences 166 TF Northeastern University 360 Huntington Avenue Boston, MA 02115 USA 617-373-3216 office 617-373-8886 fax r.loring@northeastern.edu

Hi Ralph, This is a very complicated situation because not only is it a large multimer, but also one of the structures is a fusion protein with cytochrome b562. Certainly with ChimeraX's Modeller interface, you can do two types of calculations: comparative modeling (modeling based on templates) and filling in missing segments (modeling not based on templates, aka Model Loops). Not sure you need to do any comparative modeling in this case, however, and I'm not clear on what is missing after you put the two structures together. If you were just overlapping and then wanted to make a combined model from the non-overlapped parts, that is not really a Modeller thing. You would just superimpose and then delete the redundant atoms and combine the rest into a single model. You might want to delete the cytochrome b562 insertion as well. The page for 7KOO at RCSB PDB tells me this fusion insertion is residues 353-457. Unfortunately it's a huge mess to combine the two multimers because the chain IDs aren't arranged in the same way in the two structures. These commands would open the two structures, keep only the first NMR model, delete the insertion, and superimpose (note the different orders of chain IDs in the specs): open 7rpm close #1.2-end open 7koo hide atoms show ribbons delete #2/A-E:353-457 mm #1/A,E,D,C,B to #2/A,B,C,D,E pair ss So, if you were trying to make a single atomic model for further calculations, besides simply combining (see "combine" command), it would probably also involve a fair amount of manual text-editing of PDB files. (If this is just for a schematic figure, you could just hide the overlapping parts instead of trying to make a usable single model with all this combining, however.) Say you have a model that combines these two existing structures. Are there still parts that are missing? Open the full UniProt sequence: open uniprot:acha7_human sequence associate /A-E acha7_human If there is still something missing relative to the full UniProt sequence, you would use that sequence as the target for Model Loops. With loop or missing-segment modeling using Modeller, there is no template. It just starts with the structure you already have and generally keeps it the same, except fills in the missing parts relative to the target sequence. However, this is only going to reliably model quite short segments (e.g. loops of <10 residues). It's not going to predict the structure of a whole domain, for example. If you don't have any template for the missing part, although that part will be built, it will probably look like a pile of spaghetti. That made me think, well, what about AlphaFold prediction? The feasibility of running a new calculation especially on a large multimer may be low (and can take many hours if it even finishes) but the following commands (starting with an empty session) would get the single-chain prediction 5 times and match it to the chains in 7koo: open 7koo hide atoms show ribbon delete /A-E:353-457 delete /F-J alphafold match #1/A-E trim false hide #1 models Before the alphafold fetch/match part, the above commands get rid of the fusion protein insertion and the bungarotoxin chains so that only the AchR chains are left. There will be error messages that the cytochrome model structure could not be matched, which can be ignored since you don't care about a prediction for cytochrome b-whatever anyway. AlphaFold is good at combining what is known from existing structures together, but it may or may not be able to predict the part without any similar known structures. The red/orange parts are the parts deemed unreliable or disordered, and yellow is medium. See ChimeraX alphafold help: <https://rbvi.ucsf.edu/chimerax/docs/user/commands/alphafold.html> Best, Elaine ----- Elaine C. Meng, Ph.D. UCSF Chimera(X) team Department of Pharmaceutical Chemistry University of California, San Francisco
On Feb 14, 2022, at 1:44 PM, Ralph Loring via ChimeraX-users <chimerax-users@cgl.ucsf.edu> wrote:
Hi, The intracellular domain (ICD) of Alpha7 nicotinic receptors, like almost all eukaryotic Cys-loop receptors, is not resolved using X-ray crystallography or cryo-EM. Last week the PDB for an ensemble of NMR ICD structures for the receptor was released (PDB 7RPM). I've been unsuccessfully trying to use ChimeraX and Modeller to make a model of a subunit that includes the ICD. The 7RPM models include all 4 transmembrane domains and the ICD (but lacks the extracellular domain), so there is considerable overlap with the 7KOO PDB fo a7. I started by deleting all but one of the identical subunits in the 7KOO pdb and loaded one of the 7RPM models and tried to use Modeller to make a construct that includes both the extracellular domain and the ICD. Unfortunately, even though both 7KOO and the single 7RPM are in the sequence alignments window, the ChimeraX-Modeller combination ignores the structure that is not chosen as the target (e.g. if 7RPM is the target, there is no extracellular domain, but if 7KOO is the target, there is either no ICD or the ICD that is shown bears no relevance to the structure in 7RPM). I can align the two structures well with a low RMSD using Matchmaker because of the TMDs overlapping. I just want to fill in the missing parts. Is there a special trick to get Modeller to fill in the missing parts using ChimeraX as the interface? I'm not that experienced using Modeller. Thanks, Ralph H. Loring Associate Professor of Pharmacology Department of Pharmaceutical Sciences 166 TF Northeastern University 360 Huntington Avenue Boston, MA 02115 USA 617-373-3216 office 617-373-8886 fax r.loring@northeastern.edu _______________________________________________ ChimeraX-users mailing list ChimeraX-users@cgl.ucsf.edu Manage subscription: https://www.rbvi.ucsf.edu/mailman/listinfo/chimerax-users
To clarify, the "alphafold match" command would get already-made single-chain models from the freely available AlphaFold Database. It does not run a new AlphaFold calculation. It just superimposes the single-chain predictions onto the multimer structure that was already open, which is not the same as using it to predict a multimer. However, in practice the result is often quite reasonable, if there are already experimentally known structures with the same type of multimerization. I also meant to include more help links in the previous reply... matchmaker <https://rbvi.ucsf.edu/chimerax/docs/user/commands/matchmaker.html> combine <https://rbvi.ucsf.edu/chimerax/docs/user/commands/combine.html> Model Loops <https://rbvi.ucsf.edu/chimerax/docs/user/tools/modelloops.html> Best, Elaine
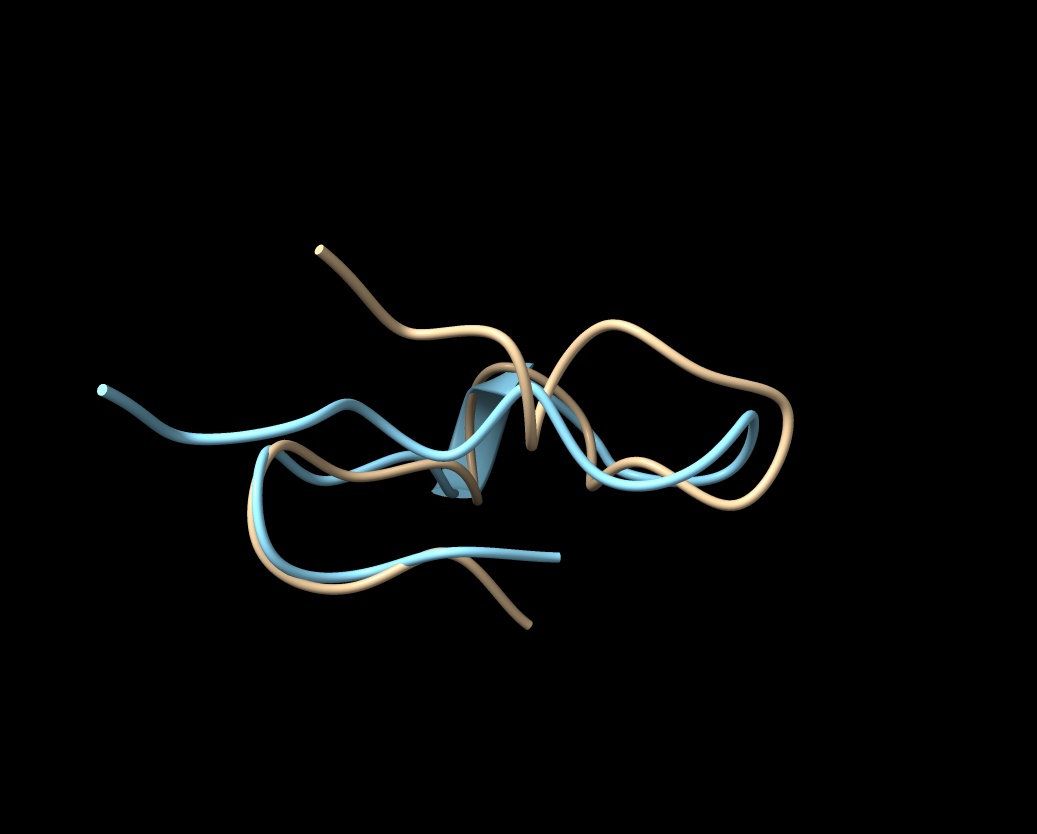
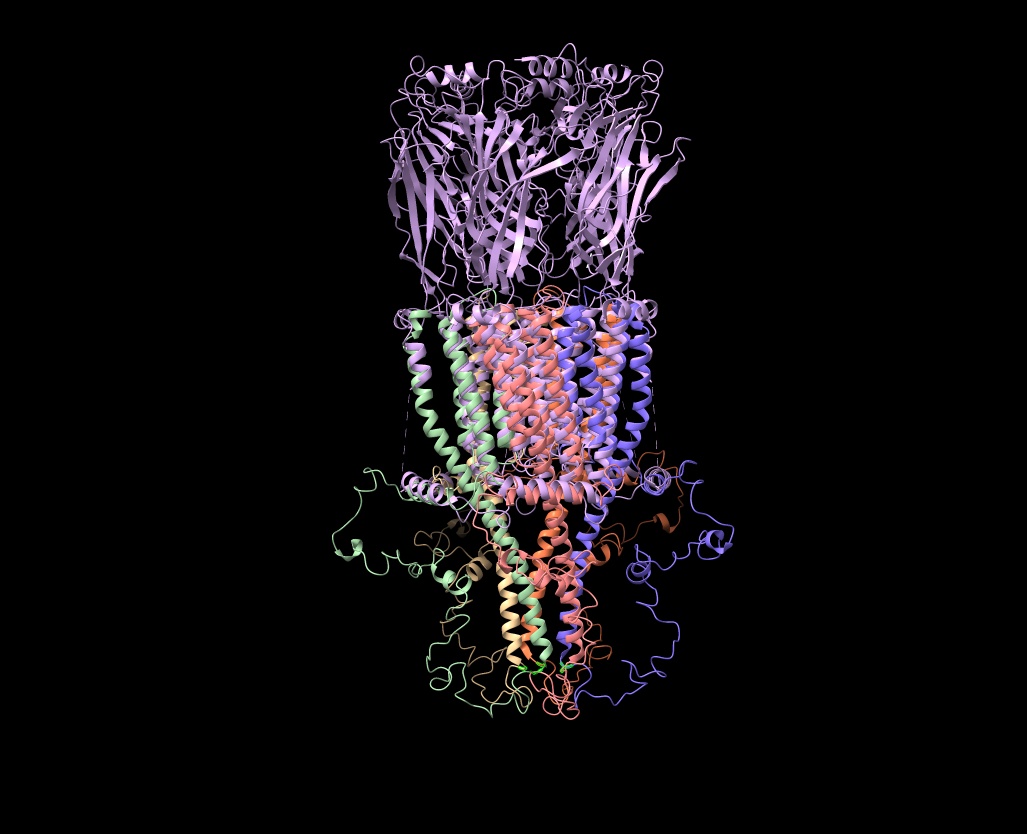
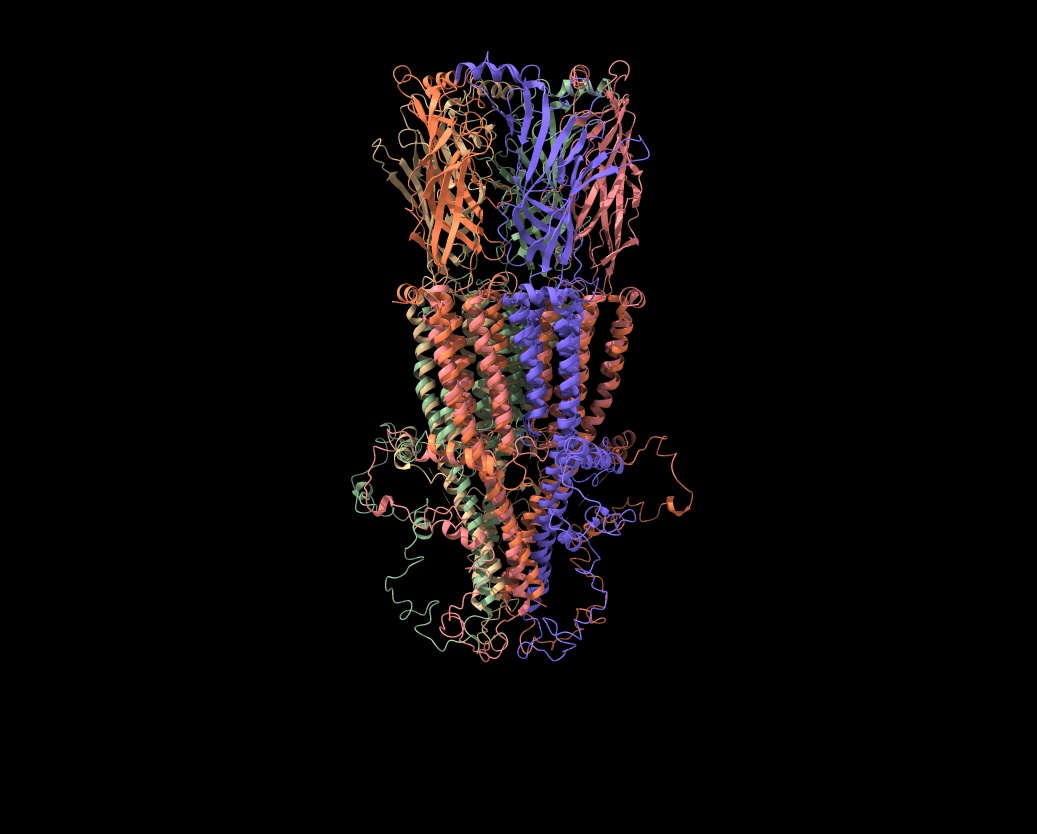
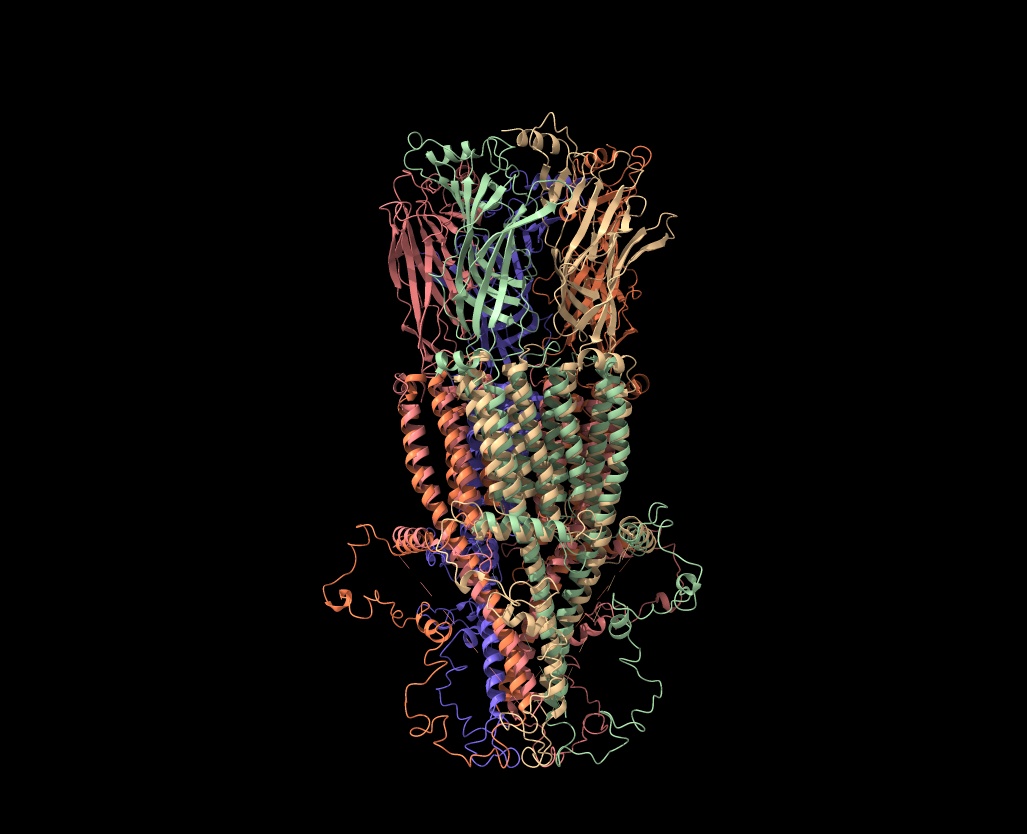
Hi Elaine Meng, Thanks so much for your suggestions! However, I’m not thrilled about the Alphafold idea, as I’ve tried to use it for receptors and keep getting booted out before the job finishes (I’d like to get Colab Pro, but I can’t figure out how to sign up with a university credit card; it’s only $0.55 per month in taxes, but my account manager will go ballistic if I pay any taxes and Google doesn’t answer my emails about how to pay with a tax exempt card). Anyway, I got Alphafold to work for a 22 amino acid mu-conotoxin (Mu-Conotoxin P0C349.pdb) and the predicted structure (in blue) wasn’t great (see attached). The RMSD between the Alphafold predicted pdb and the original (tan) was 4.1 angstroms, so not good. If there is some way to guarantee that Alphafold would use the 7RPM.pdb as a model for the intracellular loop, it might work, but the human alpha7 prediction from Alphafold before 7RPM was released (AF-P36544) is not good for the intracellular loop. There is now another Alphafold version (AF-A0A1W2PN81) that is equally bad. Your scripts were incredibly helpful, as I haven’t figured out the syntax for using the command line. Is there a tutorial on using commands in ChimeraX? (I have the list of commands in the user guide [ https://www.cgl.ucsf.edu/chimerax/docs/user/index.html#commands], but only rarely does that include examples of how the commands are used and I can't figure out the syntax code). What I’d like to do is to delete all the overlapping amino acids in either 7EKI.pdb (which has eGFP in the cytoplasmic loop instead of cytochrome) or 7KOO.pdb with one of the 7RPM ensemble and then bond the ends together to get an approximation of what the intact receptor would look like without overlaps. But I can’t get the bond command correct in a test case using the two versions of mu-contoxin (see attached csx file). I know I want to bond #1Cys22CA to #2Arg1, but I don’t even know how to specify the N-terminal nitrogen. (I can make the bond in Chimera using a combination of Select/Atom Specifier and then Tools/Structure Editing/Build Structure/Join Models, but that approach avoids using commands and I want to know how to do this in ChimeraX). I know I’m going to have to futz with phi and psi after making the bond, but I can’t even get that far (and there's two bonds to make). By the way, Matchmaker does a good job of lining up the overlapping amino acids with any combination of the receptor pdbs. As you can see, they are very comparable (the dotted lines point down rather than up in your method). But I’d like to get a pdb with a single chain for each complete subunit at some point. Thanks for all your help! Any suggestions would be most appreciated. Ralph Loring On Mon, Feb 14, 2022 at 6:33 PM Elaine Meng <meng@cgl.ucsf.edu> wrote:
To clarify, the "alphafold match" command would get already-made single-chain models from the freely available AlphaFold Database. It does not run a new AlphaFold calculation. It just superimposes the single-chain predictions onto the multimer structure that was already open, which is not the same as using it to predict a multimer. However, in practice the result is often quite reasonable, if there are already experimentally known structures with the same type of multimerization.
I also meant to include more help links in the previous reply...
matchmaker <https://rbvi.ucsf.edu/chimerax/docs/user/commands/matchmaker.html>
combine <https://rbvi.ucsf.edu/chimerax/docs/user/commands/combine.html>
Model Loops <https://rbvi.ucsf.edu/chimerax/docs/user/tools/modelloops.html>
Best, Elaine
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Hi Ralph, I wouldn't suggest running AlphaFold on a system this large. My first reply had the commands to assemble your model from the pre-existing free AlphaFold database (i.e. not running any AlphaFold calculation). I do understand that may not be quite as desired since you can't control what was used as a template, but it might be a better starting point for further structure editing since it takes care of some of the residue numbering and chain ID correspondences, at least. For reasons stated before and repeated here (stemming from multiple chains and lack of chain ID correspondence between the starting models, as well as tedium in trying to figure out what residue number ranges need to be deleted), it may be difficult to assemble a model using the workflow you had in mind. By the way, if you use any icons or menu items, the corresponding command shows up in the Log. So that is one way to learn commands. All of the tutorials contain many examples of commands as well. <https://www.rbvi.ucsf.edu/chimerax/tutorials.html> ChimeraX does not yet have a join models tool like in Chimera that simultaneously combines two models into one and bonds them while moving one model to a reasonable location. So in ChimeraX you have to position the models appropriately first (taken care of by the matchmaker commands I sent you earlier), then (if needed) delete all the extra residues (easier said than done), then combine into a single model, then add bond(s). Unfortunately the chain IDs will not match up automatically after the combine command, so for a usable a result it would probably entail writing out the single combined model as PDB, doing a lot of manual text-editing of the PDB file to fix up PDB IDs, and then reopening the file and bonding the atoms (well actually the text-editing could take care of that too). You cannot bond atoms that are in different models (#1 and #2 in your example), and I should put something about that in the "bond" documentation, although you will get an error message that says so if you try to do it. That is why you would have to use "combine" to create a single combined model first. You don't have to use command-line specification to bond the atoms with a command. If you hide ribbons and show backbone atoms you can select 2 backbone atoms (Shift-click, Shift-Ctrl-click on them in the graphics window) and then use command "bond sel reasonable false" (by default you can't see backbone atoms when the ribbon is shown). If you did want to specify in command line, as alluded to above, hovering the mouse over some atom will show its chain ID, residue number, and atom name. Usually backbone atoms are named N, CA, C, O but it may be different on the terminal residues. #1:25@ca means model 1, residue 25, atom CA. There are several examples like that in the "atom specification" page: <https://rbvi.ucsf.edu/chimerax/docs/user/commands/atomspec.html#hierarchy> "bond" <https://rbvi.ucsf.edu/chimerax/docs/user/commands/bond.html> "combine" <https://rbvi.ucsf.edu/chimerax/docs/user/commands/combine.html> It may be quite difficult/tedious to figure out what residue ranges overlap (or comes from some unrelated fusion protein), and thus what should be deleted. Keep in mind also that the numbering may not even be the same between the two structures, e.g. residue 10 in the uniprot seq might be 8 in one structure and 9 in the other. How I looked up the residue numbers of the fusion protein part of one structure before my first reply was by looking at the RCSB PDB page for that structure and mousing over the sequence view. You could also keep hovering the mouse over the structures (while hiding the others) to see balloon help to figure out where some residue number falls on the chain and writing down the ranges of residue numbers that are present in each. Personally I'd do it by looking at the RCSB PDB page and/or viewing the contents of the PDB files using a text editor, but that may be worse if you're not familiar with PDB files. Another way is to associate each structure with the uniprot sequence and then determining what's present/absent in each structure in the sequence window. I hope this helps, Elaine ----- Elaine C. Meng, Ph.D. UCSF Chimera(X) team Department of Pharmaceutical Chemistry University of California, San Francisco
On Feb 17, 2022, at 3:48 PM, Ralph Loring <rhloring@gmail.com> wrote:
Hi Elaine Meng,
Thanks so much for your suggestions! However, I’m not thrilled about the Alphafold idea, as I’ve tried to use it for receptors and keep getting booted out before the job finishes (I’d like to get Colab Pro, but I can’t figure out how to sign up with a university credit card; it’s only $0.55 per month in taxes, but my account manager will go ballistic if I pay any taxes and Google doesn’t answer my emails about how to pay with a tax exempt card). Anyway, I got Alphafold to work for a 22 amino acid mu-conotoxin (Mu-Conotoxin P0C349.pdb) and the predicted structure (in blue) wasn’t great (see attached). The RMSD between the Alphafold predicted pdb and the original (tan) was 4.1 angstroms, so not good. If there is some way to guarantee that Alphafold would use the 7RPM.pdb as a model for the intracellular loop, it might work, but the human alpha7 prediction from Alphafold before 7RPM was released (AF-P36544) is not good for the intracellular loop. There is now another Alphafold version (AF-A0A1W2PN81) that is equally bad.
Your scripts were incredibly helpful, as I haven’t figured out the syntax for using the command line. Is there a tutorial on using commands in ChimeraX? (I have the list of commands in the user guide [https://www.cgl.ucsf.edu/chimerax/docs/user/index.html#commands], but only rarely does that include examples of how the commands are used and I can't figure out the syntax code). What I’d like to do is to delete all the overlapping amino acids in either 7EKI.pdb (which has eGFP in the cytoplasmic loop instead of cytochrome) or 7KOO.pdb with one of the 7RPM ensemble and then bond the ends together to get an approximation of what the intact receptor would look like without overlaps. But I can’t get the bond command correct in a test case using the two versions of mu-contoxin (see attached csx file). I know I want to bond #1Cys22CA to #2Arg1, but I don’t even know how to specify the N-terminal nitrogen. (I can make the bond in Chimera using a combination of Select/Atom Specifier and then Tools/Structure Editing/Build Structure/Join Models, but that approach avoids using commands and I want to know how to do this in ChimeraX). I know I’m going to have to futz with phi and psi after making the bond, but I can’t even get that far (and there's two bonds to make).
By the way, Matchmaker does a good job of lining up the overlapping amino acids with any combination of the receptor pdbs. As you can see, they are very comparable (the dotted lines point down rather than up in your method). But I’d like to get a pdb with a single chain for each complete subunit at some point.
Thanks for all your help! Any suggestions would be most appreciated.
Ralph Loring
On Mon, Feb 14, 2022 at 6:33 PM Elaine Meng <meng@cgl.ucsf.edu> wrote: To clarify, the "alphafold match" command would get already-made single-chain models from the freely available AlphaFold Database. It does not run a new AlphaFold calculation. It just superimposes the single-chain predictions onto the multimer structure that was already open, which is not the same as using it to predict a multimer. However, in practice the result is often quite reasonable, if there are already experimentally known structures with the same type of multimerization.
I also meant to include more help links in the previous reply...
matchmaker <https://rbvi.ucsf.edu/chimerax/docs/user/commands/matchmaker.html>
combine <https://rbvi.ucsf.edu/chimerax/docs/user/commands/combine.html>
Model Loops <https://rbvi.ucsf.edu/chimerax/docs/user/tools/modelloops.html>
Best, Elaine
<Superimposed AF- & native Mu-conotoxins.jpg><Bond AF-mu CTX to mu-CTX.cxs><Superimposed 7KOO without BGT and 7RPM Meng's method.jpg><Superimposed 7EKI and 7RPM.jpg><Superimposed 7KOO without BGT and 7RPM.jpg>

Hi Ralph, If you are trying to run alphafold to predict the acetylcholine receptor alpha7 pentamer, that is 2500 total amino acids (500 per alpha7 monmer), then there is no way that is going to run on Google Colab which fails beyond about 1000 amino acids. (Some alphafold size tests here: https://www.rbvi.ucsf.edu/chimerax/data/alphafold-jan2022/afspeed.html) Colab Pro definitely will not help. Also all Google Colab AlphaFold scripts including what ChimeraX uses run a limited alphafold with no structure templates. See the main AlphaFold Google Colab page for more details https://colab.research.google.com/github/deepmind/alphafold/blob/main/notebo... If you could install and run full AlphaFold on say an Nvidia RTX 3090 with 24 GB of GPU memory it might succeed (not run out of memory), but it is near the size limit. You would probably need a much more exotic Nvidia A40 (48 Gbytes) or A100 or similar. And even if you run that, you are expecting too much if you think it is going to give you a fully accurate prediction -- AlphaFold produces lots of poor models for large structures. One more reality check on the capabilities of AlphaFold. It has no option to tell it exactly what PDB templates you want to use -- it chooses the "best 4", I have never seen the criteria documented for what "best 4" means, and further the log output from AlphaFold does not even tell you which 4 templates it uses. You could probably point it to a PDB database with only 7RPM if you wanted to just use that as a template if you can setup your own AlphaFold installation. In summary, definitely don't expect miracles or ease of use from AlphaFold. Of course the hand construction of Frankenstein models described by Elaine is likewise no easy task. Tom
On Feb 17, 2022, at 3:48 PM, Ralph Loring via ChimeraX-users <chimerax-users@cgl.ucsf.edu> wrote:
Hi Elaine Meng,
Thanks so much for your suggestions! However, I’m not thrilled about the Alphafold idea, as I’ve tried to use it for receptors and keep getting booted out before the job finishes (I’d like to get Colab Pro, but I can’t figure out how to sign up with a university credit card; it’s only $0.55 per month in taxes, but my account manager will go ballistic if I pay any taxes and Google doesn’t answer my emails about how to pay with a tax exempt card). Anyway, I got Alphafold to work for a 22 amino acid mu-conotoxin (Mu-Conotoxin P0C349.pdb) and the predicted structure (in blue) wasn’t great (see attached). The RMSD between the Alphafold predicted pdb and the original (tan) was 4.1 angstroms, so not good. If there is some way to guarantee that Alphafold would use the 7RPM.pdb as a model for the intracellular loop, it might work, but the human alpha7 prediction from Alphafold before 7RPM was released (AF-P36544) is not good for the intracellular loop. There is now another Alphafold version (AF-A0A1W2PN81) that is equally bad.
Your scripts were incredibly helpful, as I haven’t figured out the syntax for using the command line. Is there a tutorial on using commands in ChimeraX? (I have the list of commands in the user guide [https://www.cgl.ucsf.edu/chimerax/docs/user/index.html#commands <https://www.cgl.ucsf.edu/chimerax/docs/user/index.html#commands>], but only rarely does that include examples of how the commands are used and I can't figure out the syntax code). What I’d like to do is to delete all the overlapping amino acids in either 7EKI.pdb (which has eGFP in the cytoplasmic loop instead of cytochrome) or 7KOO.pdb with one of the 7RPM ensemble and then bond the ends together to get an approximation of what the intact receptor would look like without overlaps. But I can’t get the bond command correct in a test case using the two versions of mu-contoxin (see attached csx file). I know I want to bond #1Cys22CA to #2Arg1, but I don’t even know how to specify the N-terminal nitrogen. (I can make the bond in Chimera using a combination of Select/Atom Specifier and then Tools/Structure Editing/Build Structure/Join Models, but that approach avoids using commands and I want to know how to do this in ChimeraX). I know I’m going to have to futz with phi and psi after making the bond, but I can’t even get that far (and there's two bonds to make).
By the way, Matchmaker does a good job of lining up the overlapping amino acids with any combination of the receptor pdbs. As you can see, they are very comparable (the dotted lines point down rather than up in your method). But I’d like to get a pdb with a single chain for each complete subunit at some point.
Thanks for all your help! Any suggestions would be most appreciated.
Ralph Loring
On Mon, Feb 14, 2022 at 6:33 PM Elaine Meng <meng@cgl.ucsf.edu <mailto:meng@cgl.ucsf.edu>> wrote: To clarify, the "alphafold match" command would get already-made single-chain models from the freely available AlphaFold Database. It does not run a new AlphaFold calculation. It just superimposes the single-chain predictions onto the multimer structure that was already open, which is not the same as using it to predict a multimer. However, in practice the result is often quite reasonable, if there are already experimentally known structures with the same type of multimerization.
I also meant to include more help links in the previous reply...
matchmaker <https://rbvi.ucsf.edu/chimerax/docs/user/commands/matchmaker.html <https://rbvi.ucsf.edu/chimerax/docs/user/commands/matchmaker.html>>
combine <https://rbvi.ucsf.edu/chimerax/docs/user/commands/combine.html <https://rbvi.ucsf.edu/chimerax/docs/user/commands/combine.html>>
Model Loops <https://rbvi.ucsf.edu/chimerax/docs/user/tools/modelloops.html <https://rbvi.ucsf.edu/chimerax/docs/user/tools/modelloops.html>>
Best, Elaine
<Superimposed AF- & native Mu-conotoxins.jpg><Bond AF-mu CTX to mu-CTX.cxs><Superimposed 7KOO without BGT and 7RPM Meng's method.jpg><Superimposed 7EKI and 7RPM.jpg><Superimposed 7KOO without BGT and 7RPM.jpg>_______________________________________________ ChimeraX-users mailing list ChimeraX-users@cgl.ucsf.edu Manage subscription: https://www.rbvi.ucsf.edu/mailman/listinfo/chimerax-users
Hi Tom, No, I was running only one subunit (502 amino acids) at a time. I'm trying other approaches with other software now to incorporate the ICD. When I bonded the different domains from multiple pdbs in Chimera, it really did turn into a Frankenstein. Also, I'd seen a youtube presentation suggesting that Modeller could handle more than one pdb, and I was hoping that would work as a way to combine the different domains. That didn't work out either. But as far as learning syntax from the log, that only works for commands that you can run from pull-down menus. All I know is that the syntax for the bond command in ChimeraX is different than the equivalent in Chimera. If someone could just give me an example of a script that picks the atoms and makes a CN peptide bond, that would be very thankful. Thanks for all your help, Ralph On Fri, Feb 18, 2022 at 1:21 AM Tom Goddard <goddard@sonic.net> wrote:
Hi Ralph,
If you are trying to run alphafold to predict the acetylcholine receptor alpha7 pentamer, that is 2500 total amino acids (500 per alpha7 monmer), then there is no way that is going to run on Google Colab which fails beyond about 1000 amino acids. (Some alphafold size tests here: https://www.rbvi.ucsf.edu/chimerax/data/alphafold-jan2022/afspeed.html) Colab Pro definitely will not help. Also all Google Colab AlphaFold scripts including what ChimeraX uses run a limited alphafold with no structure templates. See the main AlphaFold Google Colab page for more details
https://colab.research.google.com/github/deepmind/alphafold/blob/main/notebo...
If you could install and run full AlphaFold on say an Nvidia RTX 3090 with 24 GB of GPU memory it might succeed (not run out of memory), but it is near the size limit. You would probably need a much more exotic Nvidia A40 (48 Gbytes) or A100 or similar. And even if you run that, you are expecting too much if you think it is going to give you a fully accurate prediction -- AlphaFold produces lots of poor models for large structures.
One more reality check on the capabilities of AlphaFold. It has no option to tell it exactly what PDB templates you want to use -- it chooses the "best 4", I have never seen the criteria documented for what "best 4" means, and further the log output from AlphaFold does not even tell you which 4 templates it uses. You could probably point it to a PDB database with only 7RPM if you wanted to just use that as a template if you can setup your own AlphaFold installation.
In summary, definitely don't expect miracles or ease of use from AlphaFold.
Of course the hand construction of Frankenstein models described by Elaine is likewise no easy task.
Tom
On Feb 17, 2022, at 3:48 PM, Ralph Loring via ChimeraX-users < chimerax-users@cgl.ucsf.edu> wrote:
Hi Elaine Meng,
Thanks so much for your suggestions! However, I’m not thrilled about the Alphafold idea, as I’ve tried to use it for receptors and keep getting booted out before the job finishes (I’d like to get Colab Pro, but I can’t figure out how to sign up with a university credit card; it’s only $0.55 per month in taxes, but my account manager will go ballistic if I pay any taxes and Google doesn’t answer my emails about how to pay with a tax exempt card). Anyway, I got Alphafold to work for a 22 amino acid mu-conotoxin (Mu-Conotoxin P0C349.pdb) and the predicted structure (in blue) wasn’t great (see attached). The RMSD between the Alphafold predicted pdb and the original (tan) was 4.1 angstroms, so not good. If there is some way to guarantee that Alphafold would use the 7RPM.pdb as a model for the intracellular loop, it might work, but the human alpha7 prediction from Alphafold before 7RPM was released (AF-P36544) is not good for the intracellular loop. There is now another Alphafold version (AF-A0A1W2PN81) that is equally bad.
Your scripts were incredibly helpful, as I haven’t figured out the syntax for using the command line. Is there a tutorial on using commands in ChimeraX? (I have the list of commands in the user guide [ https://www.cgl.ucsf.edu/chimerax/docs/user/index.html#commands], but only rarely does that include examples of how the commands are used and I can't figure out the syntax code). What I’d like to do is to delete all the overlapping amino acids in either 7EKI.pdb (which has eGFP in the cytoplasmic loop instead of cytochrome) or 7KOO.pdb with one of the 7RPM ensemble and then bond the ends together to get an approximation of what the intact receptor would look like without overlaps. But I can’t get the bond command correct in a test case using the two versions of mu-contoxin (see attached csx file). I know I want to bond #1Cys22CA to #2Arg1, but I don’t even know how to specify the N-terminal nitrogen. (I can make the bond in Chimera using a combination of Select/Atom Specifier and then Tools/Structure Editing/Build Structure/Join Models, but that approach avoids using commands and I want to know how to do this in ChimeraX). I know I’m going to have to futz with phi and psi after making the bond, but I can’t even get that far (and there's two bonds to make).
By the way, Matchmaker does a good job of lining up the overlapping amino acids with any combination of the receptor pdbs. As you can see, they are very comparable (the dotted lines point down rather than up in your method). But I’d like to get a pdb with a single chain for each complete subunit at some point.
Thanks for all your help! Any suggestions would be most appreciated.
Ralph Loring
On Mon, Feb 14, 2022 at 6:33 PM Elaine Meng <meng@cgl.ucsf.edu> wrote:
To clarify, the "alphafold match" command would get already-made single-chain models from the freely available AlphaFold Database. It does not run a new AlphaFold calculation. It just superimposes the single-chain predictions onto the multimer structure that was already open, which is not the same as using it to predict a multimer. However, in practice the result is often quite reasonable, if there are already experimentally known structures with the same type of multimerization.
I also meant to include more help links in the previous reply...
matchmaker <https://rbvi.ucsf.edu/chimerax/docs/user/commands/matchmaker.html>
combine <https://rbvi.ucsf.edu/chimerax/docs/user/commands/combine.html>
Model Loops <https://rbvi.ucsf.edu/chimerax/docs/user/tools/modelloops.html>
Best, Elaine
<Superimposed AF- & native Mu-conotoxins.jpg><Bond AF-mu CTX to mu-CTX.cxs><Superimposed 7KOO without BGT and 7RPM Meng's method.jpg><Superimposed 7EKI and 7RPM.jpg><Superimposed 7KOO without BGT and 7RPM.jpg>
ChimeraX-users mailing list ChimeraX-users@cgl.ucsf.edu Manage subscription: https://www.rbvi.ucsf.edu/mailman/listinfo/chimerax-users
Hi Ralph, Here is an example: bond #1/A:25@C #1/A:38@N reasonable false ... to add a bond in model 1 between residue 25 in chain A, atom name C, and residue 38 in chain A, atom name N. Again, you CANNOT make a bond between atoms that are in different models. If you have the pieces in different models you have to combine them into a single model first and then form the bond within the new combined model. And of course, you have to substitute in your residue numbers, chain IDs, and atom names according to what is in your actual structure. <https://rbvi.ucsf.edu/chimerax/docs/user/commands/bond.html> <https://rbvi.ucsf.edu/chimerax/docs/user/commands/combine.html> In ChimeraX, you are allowed to give more than 2 atoms and form all pairwise bonds, but you need the "reasonable false" part if you want them even if the distances are too long for a covalent bond. If you have trouble with the command-line atom spec part, you could just select the atoms from the screen like I explained in my previous reply and then use command: bond sel reasonable false <https://rbvi.ucsf.edu/chimerax/docs/user/selection.html> I hope this helps, Elaine ----- Elaine C. Meng, Ph.D. UCSF Chimera(X) team Department of Pharmaceutical Chemistry University of California, San Francisco
On Feb 18, 2022, at 4:01 AM, Ralph Loring <rhloring@gmail.com> wrote:
Hi Tom, No, I was running only one subunit (502 amino acids) at a time. I'm trying other approaches with other software now to incorporate the ICD. When I bonded the different domains from multiple pdbs in Chimera, it really did turn into a Frankenstein. Also, I'd seen a youtube presentation suggesting that Modeller could handle more than one pdb, and I was hoping that would work as a way to combine the different domains. That didn't work out either. But as far as learning syntax from the log, that only works for commands that you can run from pull-down menus. All I know is that the syntax for the bond command in ChimeraX is different than the equivalent in Chimera. If someone could just give me an example of a script that picks the atoms and makes a CN peptide bond, that would be very thankful. Thanks for all your help, Ralph
On Fri, Feb 18, 2022 at 1:21 AM Tom Goddard <goddard@sonic.net> wrote: Hi Ralph,
If you are trying to run alphafold to predict the acetylcholine receptor alpha7 pentamer, that is 2500 total amino acids (500 per alpha7 monmer), then there is no way that is going to run on Google Colab which fails beyond about 1000 amino acids. (Some alphafold size tests here: https://www.rbvi.ucsf.edu/chimerax/data/alphafold-jan2022/afspeed.html) Colab Pro definitely will not help. Also all Google Colab AlphaFold scripts including what ChimeraX uses run a limited alphafold with no structure templates. See the main AlphaFold Google Colab page for more details
https://colab.research.google.com/github/deepmind/alphafold/blob/main/notebo...
If you could install and run full AlphaFold on say an Nvidia RTX 3090 with 24 GB of GPU memory it might succeed (not run out of memory), but it is near the size limit. You would probably need a much more exotic Nvidia A40 (48 Gbytes) or A100 or similar. And even if you run that, you are expecting too much if you think it is going to give you a fully accurate prediction -- AlphaFold produces lots of poor models for large structures.
One more reality check on the capabilities of AlphaFold. It has no option to tell it exactly what PDB templates you want to use -- it chooses the "best 4", I have never seen the criteria documented for what "best 4" means, and further the log output from AlphaFold does not even tell you which 4 templates it uses. You could probably point it to a PDB database with only 7RPM if you wanted to just use that as a template if you can setup your own AlphaFold installation.
In summary, definitely don't expect miracles or ease of use from AlphaFold.
Of course the hand construction of Frankenstein models described by Elaine is likewise no easy task.
Tom
On Feb 17, 2022, at 3:48 PM, Ralph Loring via ChimeraX-users <chimerax-users@cgl.ucsf.edu> wrote:
Hi Elaine Meng,
Thanks so much for your suggestions! However, I’m not thrilled about the Alphafold idea, as I’ve tried to use it for receptors and keep getting booted out before the job finishes (I’d like to get Colab Pro, but I can’t figure out how to sign up with a university credit card; it’s only $0.55 per month in taxes, but my account manager will go ballistic if I pay any taxes and Google doesn’t answer my emails about how to pay with a tax exempt card). Anyway, I got Alphafold to work for a 22 amino acid mu-conotoxin (Mu-Conotoxin P0C349.pdb) and the predicted structure (in blue) wasn’t great (see attached). The RMSD between the Alphafold predicted pdb and the original (tan) was 4.1 angstroms, so not good. If there is some way to guarantee that Alphafold would use the 7RPM.pdb as a model for the intracellular loop, it might work, but the human alpha7 prediction from Alphafold before 7RPM was released (AF-P36544) is not good for the intracellular loop. There is now another Alphafold version (AF-A0A1W2PN81) that is equally bad.
Your scripts were incredibly helpful, as I haven’t figured out the syntax for using the command line. Is there a tutorial on using commands in ChimeraX? (I have the list of commands in the user guide [https://www.cgl.ucsf.edu/chimerax/docs/user/index.html#commands], but only rarely does that include examples of how the commands are used and I can't figure out the syntax code). What I’d like to do is to delete all the overlapping amino acids in either 7EKI.pdb (which has eGFP in the cytoplasmic loop instead of cytochrome) or 7KOO.pdb with one of the 7RPM ensemble and then bond the ends together to get an approximation of what the intact receptor would look like without overlaps. But I can’t get the bond command correct in a test case using the two versions of mu-contoxin (see attached csx file). I know I want to bond #1Cys22CA to #2Arg1, but I don’t even know how to specify the N-terminal nitrogen. (I can make the bond in Chimera using a combination of Select/Atom Specifier and then Tools/Structure Editing/Build Structure/Join Models, but that approach avoids using commands and I want to know how to do this in ChimeraX). I know I’m going to have to futz with phi and psi after making the bond, but I can’t even get that far (and there's two bonds to make).
By the way, Matchmaker does a good job of lining up the overlapping amino acids with any combination of the receptor pdbs. As you can see, they are very comparable (the dotted lines point down rather than up in your method). But I’d like to get a pdb with a single chain for each complete subunit at some point.
Thanks for all your help! Any suggestions would be most appreciated.
Ralph Loring
On Mon, Feb 14, 2022 at 6:33 PM Elaine Meng <meng@cgl.ucsf.edu> wrote: To clarify, the "alphafold match" command would get already-made single-chain models from the freely available AlphaFold Database. It does not run a new AlphaFold calculation. It just superimposes the single-chain predictions onto the multimer structure that was already open, which is not the same as using it to predict a multimer. However, in practice the result is often quite reasonable, if there are already experimentally known structures with the same type of multimerization.
I also meant to include more help links in the previous reply...
matchmaker <https://rbvi.ucsf.edu/chimerax/docs/user/commands/matchmaker.html>
combine <https://rbvi.ucsf.edu/chimerax/docs/user/commands/combine.html>
Model Loops <https://rbvi.ucsf.edu/chimerax/docs/user/tools/modelloops.html>
Best, Elaine
<Superimposed AF- & native Mu-conotoxins.jpg><Bond AF-mu CTX to mu-CTX.cxs><Superimposed 7KOO without BGT and 7RPM Meng's method.jpg><Superimposed 7EKI and 7RPM.jpg><Superimposed 7KOO without BGT and 7RPM.jpg>_______________________________________________ ChimeraX-users mailing list ChimeraX-users@cgl.ucsf.edu Manage subscription: https://www.rbvi.ucsf.edu/mailman/listinfo/chimerax-users
Thanks Elaine! It worked and formed 1 chain out of 2! I had not combined before trying to bond previously. Oddly, I don't see in the bond command description where that is mentioned. Also, I surmise that the "reasonable" modifier is not optional because when I omitted it, it didn't give an error, but said 0 bonds were formed. Ralph On Fri, Feb 18, 2022 at 11:45 AM Elaine Meng <meng@cgl.ucsf.edu> wrote:
Hi Ralph, Here is an example:
bond #1/A:25@C #1/A:38@N reasonable false
... to add a bond in model 1 between residue 25 in chain A, atom name C, and residue 38 in chain A, atom name N.
Again, you CANNOT make a bond between atoms that are in different models. If you have the pieces in different models you have to combine them into a single model first and then form the bond within the new combined model. And of course, you have to substitute in your residue numbers, chain IDs, and atom names according to what is in your actual structure.
<https://rbvi.ucsf.edu/chimerax/docs/user/commands/bond.html> <https://rbvi.ucsf.edu/chimerax/docs/user/commands/combine.html>
In ChimeraX, you are allowed to give more than 2 atoms and form all pairwise bonds, but you need the "reasonable false" part if you want them even if the distances are too long for a covalent bond.
If you have trouble with the command-line atom spec part, you could just select the atoms from the screen like I explained in my previous reply and then use command:
bond sel reasonable false
<https://rbvi.ucsf.edu/chimerax/docs/user/selection.html>
I hope this helps, Elaine ----- Elaine C. Meng, Ph.D. UCSF Chimera(X) team Department of Pharmaceutical Chemistry University of California, San Francisco
On Feb 18, 2022, at 4:01 AM, Ralph Loring <rhloring@gmail.com> wrote:
Hi Tom, No, I was running only one subunit (502 amino acids) at a time. I'm trying other approaches with other software now to incorporate the ICD. When I bonded the different domains from multiple pdbs in Chimera, it really did turn into a Frankenstein. Also, I'd seen a youtube presentation suggesting that Modeller could handle more than one pdb, and I was hoping that would work as a way to combine the different domains. That didn't work out either. But as far as learning syntax from the log, that only works for commands that you can run from pull-down menus. All I know is that the syntax for the bond command in ChimeraX is different than the equivalent in Chimera. If someone could just give me an example of a script that picks the atoms and makes a CN peptide bond, that would be very thankful. Thanks for all your help, Ralph
On Fri, Feb 18, 2022 at 1:21 AM Tom Goddard <goddard@sonic.net> wrote: Hi Ralph,
If you are trying to run alphafold to predict the acetylcholine receptor alpha7 pentamer, that is 2500 total amino acids (500 per alpha7 monmer), then there is no way that is going to run on Google Colab which fails beyond about 1000 amino acids. (Some alphafold size tests here: https://www.rbvi.ucsf.edu/chimerax/data/alphafold-jan2022/afspeed.html) Colab Pro definitely will not help. Also all Google Colab AlphaFold scripts including what ChimeraX uses run a limited alphafold with no structure templates. See the main AlphaFold Google Colab page for more details
https://colab.research.google.com/github/deepmind/alphafold/blob/main/notebo...
If you could install and run full AlphaFold on say an Nvidia RTX 3090
with 24 GB of GPU memory it might succeed (not run out of memory), but it is near the size limit. You would probably need a much more exotic Nvidia A40 (48 Gbytes) or A100 or similar. And even if you run that, you are expecting too much if you think it is going to give you a fully accurate prediction -- AlphaFold produces lots of poor models for large structures.
One more reality check on the capabilities of AlphaFold. It has no
option to tell it exactly what PDB templates you want to use -- it chooses the "best 4", I have never seen the criteria documented for what "best 4" means, and further the log output from AlphaFold does not even tell you which 4 templates it uses. You could probably point it to a PDB database with only 7RPM if you wanted to just use that as a template if you can setup your own AlphaFold installation.
In summary, definitely don't expect miracles or ease of use from
AlphaFold.
Of course the hand construction of Frankenstein models described by
Elaine is likewise no easy task.
Tom
On Feb 17, 2022, at 3:48 PM, Ralph Loring via ChimeraX-users <
chimerax-users@cgl.ucsf.edu> wrote:
Hi Elaine Meng,
Thanks so much for your suggestions! However, I’m not thrilled about
the Alphafold idea, as I’ve tried to use it for receptors and keep getting booted out before the job finishes (I’d like to get Colab Pro, but I can’t figure out how to sign up with a university credit card; it’s only $0.55 per month in taxes, but my account manager will go ballistic if I pay any taxes and Google doesn’t answer my emails about how to pay with a tax exempt card). Anyway, I got Alphafold to work for a 22 amino acid mu-conotoxin (Mu-Conotoxin P0C349.pdb) and the predicted structure (in blue) wasn’t great (see attached). The RMSD between the Alphafold predicted pdb and the original (tan) was 4.1 angstroms, so not good. If there is some way to guarantee that Alphafold would use the 7RPM.pdb as a model for the intracellular loop, it might work, but the human alpha7 prediction from Alphafold before 7RPM was released (AF-P36544) is not good for the intracellular loop. There is now another Alphafold version (AF-A0A1W2PN81) that is equally bad.
Your scripts were incredibly helpful, as I haven’t figured out the
syntax for using the command line. Is there a tutorial on using commands in ChimeraX? (I have the list of commands in the user guide [ https://www.cgl.ucsf.edu/chimerax/docs/user/index.html#commands], but only rarely does that include examples of how the commands are used and I can't figure out the syntax code). What I’d like to do is to delete all the overlapping amino acids in either 7EKI.pdb (which has eGFP in the cytoplasmic loop instead of cytochrome) or 7KOO.pdb with one of the 7RPM ensemble and then bond the ends together to get an approximation of what the intact receptor would look like without overlaps. But I can’t get the bond command correct in a test case using the two versions of mu-contoxin (see attached csx file). I know I want to bond #1Cys22CA to #2Arg1, but I don’t even know how to specify the N-terminal nitrogen. (I can make the bond in Chimera using a combination of Select/Atom Specifier and then Tools/Structure Editing/Build Structure/Join Models, but that approach avoids using commands and I want to know how to do this in ChimeraX). I know I’m going to have to futz with phi and psi after making the bond, but I can’t even get that far (and there's two bonds to make).
By the way, Matchmaker does a good job of lining up the overlapping
amino acids with any combination of the receptor pdbs. As you can see, they are very comparable (the dotted lines point down rather than up in your method). But I’d like to get a pdb with a single chain for each complete subunit at some point.
Thanks for all your help! Any suggestions would be most appreciated.
Ralph Loring
On Mon, Feb 14, 2022 at 6:33 PM Elaine Meng <meng@cgl.ucsf.edu> wrote: To clarify, the "alphafold match" command would get already-made
single-chain models from the freely available AlphaFold Database. It does not run a new AlphaFold calculation. It just superimposes the single-chain predictions onto the multimer structure that was already open, which is not the same as using it to predict a multimer. However, in practice the result is often quite reasonable, if there are already experimentally known structures with the same type of multimerization.
I also meant to include more help links in the previous reply...
matchmaker <https://rbvi.ucsf.edu/chimerax/docs/user/commands/matchmaker.html>
combine <https://rbvi.ucsf.edu/chimerax/docs/user/commands/combine.html>
Model Loops <https://rbvi.ucsf.edu/chimerax/docs/user/tools/modelloops.html>
Best, Elaine
<Superimposed AF- & native Mu-conotoxins.jpg><Bond AF-mu CTX to
mu-CTX.cxs><Superimposed 7KOO without BGT and 7RPM Meng's method.jpg><Superimposed 7EKI and 7RPM.jpg><Superimposed 7KOO without BGT and 7RPM.jpg>_______________________________________________
ChimeraX-users mailing list ChimeraX-users@cgl.ucsf.edu Manage subscription: https://www.rbvi.ucsf.edu/mailman/listinfo/chimerax-users
Hi Ralph, I hadn't mentioned the same-model issue (yet) in the "bond" manual page, but I've added it now, to show up in a day or two on the website version. However, when you try to bond two different models, there should already have been an error message telling you that that was the problem. As explained below and in the bond manual page, "reasonable false" forms the bond(s) even when they are deemed unrealistically long. So it is optional if the bond being formed is a normal distance (i.e. "reasonable true" is the default), but not optional if you actually want to add bond(s) between atoms that are farther apart than what is deemed a reasonable bonding distance. <https://rbvi.ucsf.edu/chimerax/docs/user/commands/bond.html> Best, Elaine
On Feb 18, 2022, at 2:31 PM, Ralph Loring via ChimeraX-users <chimerax-users@cgl.ucsf.edu> wrote:
Thanks Elaine! It worked and formed 1 chain out of 2! I had not combined before trying to bond previously. Oddly, I don't see in the bond command description where that is mentioned. Also, I surmise that the "reasonable" modifier is not optional because when I omitted it, it didn't give an error, but said 0 bonds were formed. Ralph
On Fri, Feb 18, 2022 at 11:45 AM Elaine Meng <meng@cgl.ucsf.edu> wrote: Hi Ralph, Here is an example:
bond #1/A:25@C #1/A:38@N reasonable false
... to add a bond in model 1 between residue 25 in chain A, atom name C, and residue 38 in chain A, atom name N.
Again, you CANNOT make a bond between atoms that are in different models. If you have the pieces in different models you have to combine them into a single model first and then form the bond within the new combined model. And of course, you have to substitute in your residue numbers, chain IDs, and atom names according to what is in your actual structure.
<https://rbvi.ucsf.edu/chimerax/docs/user/commands/bond.html> <https://rbvi.ucsf.edu/chimerax/docs/user/commands/combine.html>
In ChimeraX, you are allowed to give more than 2 atoms and form all pairwise bonds, but you need the "reasonable false" part if you want them even if the distances are too long for a covalent bond.
If you have trouble with the command-line atom spec part, you could just select the atoms from the screen like I explained in my previous reply and then use command:
bond sel reasonable false
<https://rbvi.ucsf.edu/chimerax/docs/user/selection.html>
I hope this helps, Elaine ----- Elaine C. Meng, Ph.D. UCSF Chimera(X) team Department of Pharmaceutical Chemistry University of California, San Francisco
On Feb 18, 2022, at 4:01 AM, Ralph Loring <rhloring@gmail.com> wrote:
Hi Tom, No, I was running only one subunit (502 amino acids) at a time. I'm trying other approaches with other software now to incorporate the ICD. When I bonded the different domains from multiple pdbs in Chimera, it really did turn into a Frankenstein. Also, I'd seen a youtube presentation suggesting that Modeller could handle more than one pdb, and I was hoping that would work as a way to combine the different domains. That didn't work out either. But as far as learning syntax from the log, that only works for commands that you can run from pull-down menus. All I know is that the syntax for the bond command in ChimeraX is different than the equivalent in Chimera. If someone could just give me an example of a script that picks the atoms and makes a CN peptide bond, that would be very thankful. Thanks for all your help, Ralph
On Fri, Feb 18, 2022 at 1:21 AM Tom Goddard <goddard@sonic.net> wrote: Hi Ralph,
If you are trying to run alphafold to predict the acetylcholine receptor alpha7 pentamer, that is 2500 total amino acids (500 per alpha7 monmer), then there is no way that is going to run on Google Colab which fails beyond about 1000 amino acids. (Some alphafold size tests here: https://www.rbvi.ucsf.edu/chimerax/data/alphafold-jan2022/afspeed.html) Colab Pro definitely will not help. Also all Google Colab AlphaFold scripts including what ChimeraX uses run a limited alphafold with no structure templates. See the main AlphaFold Google Colab page for more details
https://colab.research.google.com/github/deepmind/alphafold/blob/main/notebo...
If you could install and run full AlphaFold on say an Nvidia RTX 3090 with 24 GB of GPU memory it might succeed (not run out of memory), but it is near the size limit. You would probably need a much more exotic Nvidia A40 (48 Gbytes) or A100 or similar. And even if you run that, you are expecting too much if you think it is going to give you a fully accurate prediction -- AlphaFold produces lots of poor models for large structures.
One more reality check on the capabilities of AlphaFold. It has no option to tell it exactly what PDB templates you want to use -- it chooses the "best 4", I have never seen the criteria documented for what "best 4" means, and further the log output from AlphaFold does not even tell you which 4 templates it uses. You could probably point it to a PDB database with only 7RPM if you wanted to just use that as a template if you can setup your own AlphaFold installation.
In summary, definitely don't expect miracles or ease of use from AlphaFold.
Of course the hand construction of Frankenstein models described by Elaine is likewise no easy task.
Tom
On Feb 17, 2022, at 3:48 PM, Ralph Loring via ChimeraX-users <chimerax-users@cgl.ucsf.edu> wrote:
Hi Elaine Meng,
Thanks so much for your suggestions! However, I’m not thrilled about the Alphafold idea, as I’ve tried to use it for receptors and keep getting booted out before the job finishes (I’d like to get Colab Pro, but I can’t figure out how to sign up with a university credit card; it’s only $0.55 per month in taxes, but my account manager will go ballistic if I pay any taxes and Google doesn’t answer my emails about how to pay with a tax exempt card). Anyway, I got Alphafold to work for a 22 amino acid mu-conotoxin (Mu-Conotoxin P0C349.pdb) and the predicted structure (in blue) wasn’t great (see attached). The RMSD between the Alphafold predicted pdb and the original (tan) was 4.1 angstroms, so not good. If there is some way to guarantee that Alphafold would use the 7RPM.pdb as a model for the intracellular loop, it might work, but the human alpha7 prediction from Alphafold before 7RPM was released (AF-P36544) is not good for the intracellular loop. There is now another Alphafold version (AF-A0A1W2PN81) that is equally bad.
Your scripts were incredibly helpful, as I haven’t figured out the syntax for using the command line. Is there a tutorial on using commands in ChimeraX? (I have the list of commands in the user guide [https://www.cgl.ucsf.edu/chimerax/docs/user/index.html#commands], but only rarely does that include examples of how the commands are used and I can't figure out the syntax code). What I’d like to do is to delete all the overlapping amino acids in either 7EKI.pdb (which has eGFP in the cytoplasmic loop instead of cytochrome) or 7KOO.pdb with one of the 7RPM ensemble and then bond the ends together to get an approximation of what the intact receptor would look like without overlaps. But I can’t get the bond command correct in a test case using the two versions of mu-contoxin (see attached csx file). I know I want to bond #1Cys22CA to #2Arg1, but I don’t even know how to specify the N-terminal nitrogen. (I can make the bond in Chimera using a combination of Select/Atom Specifier and then Tools/Structure Editing/Build Structure/Join Models, but that approach avoids using commands and I want to know how to do this in ChimeraX). I know I’m going to have to futz with phi and psi after making the bond, but I can’t even get that far (and there's two bonds to make).
By the way, Matchmaker does a good job of lining up the overlapping amino acids with any combination of the receptor pdbs. As you can see, they are very comparable (the dotted lines point down rather than up in your method). But I’d like to get a pdb with a single chain for each complete subunit at some point.
Thanks for all your help! Any suggestions would be most appreciated.
Ralph Loring
On Mon, Feb 14, 2022 at 6:33 PM Elaine Meng <meng@cgl.ucsf.edu> wrote: To clarify, the "alphafold match" command would get already-made single-chain models from the freely available AlphaFold Database. It does not run a new AlphaFold calculation. It just superimposes the single-chain predictions onto the multimer structure that was already open, which is not the same as using it to predict a multimer. However, in practice the result is often quite reasonable, if there are already experimentally known structures with the same type of multimerization.
I also meant to include more help links in the previous reply...
matchmaker <https://rbvi.ucsf.edu/chimerax/docs/user/commands/matchmaker.html>
combine <https://rbvi.ucsf.edu/chimerax/docs/user/commands/combine.html>
Model Loops <https://rbvi.ucsf.edu/chimerax/docs/user/tools/modelloops.html>
Best, Elaine
<Superimposed AF- & native Mu-conotoxins.jpg><Bond AF-mu CTX to mu-CTX.cxs><Superimposed 7KOO without BGT and 7RPM Meng's method.jpg><Superimposed 7EKI and 7RPM.jpg><Superimposed 7KOO without BGT and 7RPM.jpg>_______________________________________________ ChimeraX-users mailing list ChimeraX-users@cgl.ucsf.edu Manage subscription: https://www.rbvi.ucsf.edu/mailman/listinfo/chimerax-users
_______________________________________________ ChimeraX-users mailing list ChimeraX-users@cgl.ucsf.edu Manage subscription: https://www.rbvi.ucsf.edu/mailman/listinfo/chimerax-users
Hi Ralph, 500 amino acids should be no problem for AlphaFold on free Google Colab -- I have (almost) never seen it fail on a sequence that short in runs on 100 different sequences. But if you predict a monomer you cannot expect it to position the domains and connecting loops reasonably since that is set by the packing of the multimer. Still, miraculously AlphaFold sometimes does predict monomers in exactly the right conformation even when it is intertwined with other proteins in a complex and even without structure templates (no structure templates are used with Google Colab AlphaFold). The most recent example I ran a few days ago had AlphaFold predict a monomer encapsulin-ferritin (5n5f) completely correctly for the decamer complex, baffling! (Shown at the bottom of this page https://www.rbvi.ucsf.edu/chimerax/data/vr-nih-feb2022/ferritin.html). Tom
On Feb 18, 2022, at 4:01 AM, Ralph Loring via ChimeraX-users <chimerax-users@cgl.ucsf.edu> wrote:
Hi Tom, No, I was running only one subunit (502 amino acids) at a time. I'm trying other approaches with other software now to incorporate the ICD. When I bonded the different domains from multiple pdbs in Chimera, it really did turn into a Frankenstein. Also, I'd seen a youtube presentation suggesting that Modeller could handle more than one pdb, and I was hoping that would work as a way to combine the different domains. That didn't work out either. But as far as learning syntax from the log, that only works for commands that you can run from pull-down menus. All I know is that the syntax for the bond command in ChimeraX is different than the equivalent in Chimera. If someone could just give me an example of a script that picks the atoms and makes a CN peptide bond, that would be very thankful. Thanks for all your help, Ralph
On Fri, Feb 18, 2022 at 1:21 AM Tom Goddard <goddard@sonic.net <mailto:goddard@sonic.net>> wrote: Hi Ralph,
If you are trying to run alphafold to predict the acetylcholine receptor alpha7 pentamer, that is 2500 total amino acids (500 per alpha7 monmer), then there is no way that is going to run on Google Colab which fails beyond about 1000 amino acids. (Some alphafold size tests here: https://www.rbvi.ucsf.edu/chimerax/data/alphafold-jan2022/afspeed.html <https://www.rbvi.ucsf.edu/chimerax/data/alphafold-jan2022/afspeed.html>) Colab Pro definitely will not help. Also all Google Colab AlphaFold scripts including what ChimeraX uses run a limited alphafold with no structure templates. See the main AlphaFold Google Colab page for more details
https://colab.research.google.com/github/deepmind/alphafold/blob/main/notebo... <https://colab.research.google.com/github/deepmind/alphafold/blob/main/notebo...>
If you could install and run full AlphaFold on say an Nvidia RTX 3090 with 24 GB of GPU memory it might succeed (not run out of memory), but it is near the size limit. You would probably need a much more exotic Nvidia A40 (48 Gbytes) or A100 or similar. And even if you run that, you are expecting too much if you think it is going to give you a fully accurate prediction -- AlphaFold produces lots of poor models for large structures.
One more reality check on the capabilities of AlphaFold. It has no option to tell it exactly what PDB templates you want to use -- it chooses the "best 4", I have never seen the criteria documented for what "best 4" means, and further the log output from AlphaFold does not even tell you which 4 templates it uses. You could probably point it to a PDB database with only 7RPM if you wanted to just use that as a template if you can setup your own AlphaFold installation.
In summary, definitely don't expect miracles or ease of use from AlphaFold.
Of course the hand construction of Frankenstein models described by Elaine is likewise no easy task.
Tom
On Feb 17, 2022, at 3:48 PM, Ralph Loring via ChimeraX-users <chimerax-users@cgl.ucsf.edu <mailto:chimerax-users@cgl.ucsf.edu>> wrote:
Hi Elaine Meng,
Thanks so much for your suggestions! However, I’m not thrilled about the Alphafold idea, as I’ve tried to use it for receptors and keep getting booted out before the job finishes (I’d like to get Colab Pro, but I can’t figure out how to sign up with a university credit card; it’s only $0.55 per month in taxes, but my account manager will go ballistic if I pay any taxes and Google doesn’t answer my emails about how to pay with a tax exempt card). Anyway, I got Alphafold to work for a 22 amino acid mu-conotoxin (Mu-Conotoxin P0C349.pdb) and the predicted structure (in blue) wasn’t great (see attached). The RMSD between the Alphafold predicted pdb and the original (tan) was 4.1 angstroms, so not good. If there is some way to guarantee that Alphafold would use the 7RPM.pdb as a model for the intracellular loop, it might work, but the human alpha7 prediction from Alphafold before 7RPM was released (AF-P36544) is not good for the intracellular loop. There is now another Alphafold version (AF-A0A1W2PN81) that is equally bad.
Your scripts were incredibly helpful, as I haven’t figured out the syntax for using the command line. Is there a tutorial on using commands in ChimeraX? (I have the list of commands in the user guide [https://www.cgl.ucsf.edu/chimerax/docs/user/index.html#commands <https://www.cgl.ucsf.edu/chimerax/docs/user/index.html#commands>], but only rarely does that include examples of how the commands are used and I can't figure out the syntax code). What I’d like to do is to delete all the overlapping amino acids in either 7EKI.pdb (which has eGFP in the cytoplasmic loop instead of cytochrome) or 7KOO.pdb with one of the 7RPM ensemble and then bond the ends together to get an approximation of what the intact receptor would look like without overlaps. But I can’t get the bond command correct in a test case using the two versions of mu-contoxin (see attached csx file). I know I want to bond #1Cys22CA to #2Arg1, but I don’t even know how to specify the N-terminal nitrogen. (I can make the bond in Chimera using a combination of Select/Atom Specifier and then Tools/Structure Editing/Build Structure/Join Models, but that approach avoids using commands and I want to know how to do this in ChimeraX). I know I’m going to have to futz with phi and psi after making the bond, but I can’t even get that far (and there's two bonds to make).
By the way, Matchmaker does a good job of lining up the overlapping amino acids with any combination of the receptor pdbs. As you can see, they are very comparable (the dotted lines point down rather than up in your method). But I’d like to get a pdb with a single chain for each complete subunit at some point.
Thanks for all your help! Any suggestions would be most appreciated.
Ralph Loring
On Mon, Feb 14, 2022 at 6:33 PM Elaine Meng <meng@cgl.ucsf.edu <mailto:meng@cgl.ucsf.edu>> wrote: To clarify, the "alphafold match" command would get already-made single-chain models from the freely available AlphaFold Database. It does not run a new AlphaFold calculation. It just superimposes the single-chain predictions onto the multimer structure that was already open, which is not the same as using it to predict a multimer. However, in practice the result is often quite reasonable, if there are already experimentally known structures with the same type of multimerization.
I also meant to include more help links in the previous reply...
matchmaker <https://rbvi.ucsf.edu/chimerax/docs/user/commands/matchmaker.html <https://rbvi.ucsf.edu/chimerax/docs/user/commands/matchmaker.html>>
combine <https://rbvi.ucsf.edu/chimerax/docs/user/commands/combine.html <https://rbvi.ucsf.edu/chimerax/docs/user/commands/combine.html>>
Model Loops <https://rbvi.ucsf.edu/chimerax/docs/user/tools/modelloops.html <https://rbvi.ucsf.edu/chimerax/docs/user/tools/modelloops.html>>
Best, Elaine
<Superimposed AF- & native Mu-conotoxins.jpg><Bond AF-mu CTX to mu-CTX.cxs><Superimposed 7KOO without BGT and 7RPM Meng's method.jpg><Superimposed 7EKI and 7RPM.jpg><Superimposed 7KOO without BGT and 7RPM.jpg>_______________________________________________ ChimeraX-users mailing list ChimeraX-users@cgl.ucsf.edu <mailto:ChimeraX-users@cgl.ucsf.edu> Manage subscription: https://www.rbvi.ucsf.edu/mailman/listinfo/chimerax-users <https://www.rbvi.ucsf.edu/mailman/listinfo/chimerax-users>
_______________________________________________ ChimeraX-users mailing list ChimeraX-users@cgl.ucsf.edu Manage subscription: https://www.rbvi.ucsf.edu/mailman/listinfo/chimerax-users
participants (3)
-
 Elaine Meng
Elaine Meng -
 Ralph Loring
Ralph Loring -
 Tom Goddard
Tom Goddard