
Begin forwarded message:
From: "Azad, Roksana" <razad@gc.cuny.edu> Subject: Re: [ChimeraX] #4622: save dialog: 'SaveModelOptionWidget' object has no attribute '_name' Date: May 11, 2021 at 9:00:00 AM PDT To: "ChimeraX-bugs@cgl.ucsf.edu" <ChimeraX-bugs@cgl.ucsf.edu>
Dear Eric, Thank you so much for your reply.
I have a model of my protein in pdb format, which I am trying to save as .mrc or any sort of volume file on chimera. However, since its a model and do not have any coordinate in the file like xtal or NMR structure, it won’t save it as .mrc or volume file and was giving me the error.
I want to create a volume file from the model to load on cryoSPARC or relion and create 2D projections to help me pick better 2D classes from my raw data. Do you think there is an option to do something like this on chimera?
Thank you again for your time and help in advance – very much appreciated.
Sincerely, Roksana
On May 11, 2021, at 11:50 AM, ChimeraX <ChimeraX-bugs-admin@cgl.ucsf.edu> wrote:
#4622: save dialog: 'SaveModelOptionWidget' object has no attribute '_name' -----------------------------------+---------------------------- Reporter: razad@… | Owner: Eric Pettersen Type: defect | Status: closed Priority: normal | Milestone: Component: Input/Output | Version: Resolution: duplicate | Keywords: Blocked By: | Blocking: Notify when closed: | Platform: all Project: ChimeraX | -----------------------------------+---------------------------- Changes (by Eric Pettersen):
* status: accepted => closed * resolution: => duplicate
Comment:
Hi Roksana, Thanks for reporting this problem. ChimeraX was trying to tell you that you hadn't selected anything to save in the save dialog, but there was an error in the code that generates that message, which is what you saw. So the error is easy to avoid. Nonetheless, if you want to always avoid it in the future you could use the current daily build or the 1.2.2 release candidate, both of which have this problem fixed.
--Eric
Eric Pettersen UCSF Computer Graphics Lab
-- Ticket URL: <https://urldefense.proofpoint.com/v2/url?u=https-3A__plato.cgl.ucsf.edu_trac... > ChimeraX <https://urldefense.proofpoint.com/v2/url?u=http-3A__www.rbvi.ucsf.edu_chimer... > ChimeraX Issue Tracker

Dear Roksana, The command "molmap" creates a map model from an atomic model, for example open 1zik molmap protein 8 See the "molmap" help for explanation of the resolution value and other command options: <https://rbvi.ucsf.edu/chimerax/docs/user/commands/molmap.html> I hope this helps, Elaine ----- Elaine C. Meng, Ph.D. UCSF Chimera(X) team Department of Pharmaceutical Chemistry University of California, San Francisco
Begin forwarded message:
From: "Azad, Roksana" <razad@gc.cuny.edu> Subject: Re: [ChimeraX] #4622: save dialog: 'SaveModelOptionWidget' object has no attribute '_name' Date: May 11, 2021 at 9:00:00 AM PDT To: "ChimeraX-bugs@cgl.ucsf.edu" <ChimeraX-bugs@cgl.ucsf.edu>
Dear Eric, Thank you so much for your reply.
I have a model of my protein in pdb format, which I am trying to save as .mrc or any sort of volume file on chimera. However, since its a model and do not have any coordinate in the file like xtal or NMR structure, it won’t save it as .mrc or volume file and was giving me the error.
I want to create a volume file from the model to load on cryoSPARC or relion and create 2D projections to help me pick better 2D classes from my raw data. Do you think there is an option to do something like this on chimera?
Thank you again for your time and help in advance – very much appreciated. Sincerely, Roksana

Dear Elaine, I have tried the molmap command in the past, but it shows this error "Missing or invalid "atoms" argument: invalid atoms specifier”. Just to clarify, my protein structure is a model generated from I-tasser, it doesn’t have a resolution. I was thinking that I am getting this error because there are no resolution coordinates in the file?! Thank you again for your help – very much apppreciated. Sincerely, Roksana
On May 11, 2021, at 12:09 PM, Elaine Meng <meng@cgl.ucsf.edu> wrote:
Dear Roksana, The command "molmap" creates a map model from an atomic model, for example
open 1zik molmap protein 8
See the "molmap" help for explanation of the resolution value and other command options: <https://urldefense.proofpoint.com/v2/url?u=https-3A__rbvi.ucsf.edu_chimerax_... >
I hope this helps, Elaine ----- Elaine C. Meng, Ph.D. UCSF Chimera(X) team Department of Pharmaceutical Chemistry University of California, San Francisco
Begin forwarded message:
From: "Azad, Roksana" <razad@gc.cuny.edu> Subject: Re: [ChimeraX] #4622: save dialog: 'SaveModelOptionWidget' object has no attribute '_name' Date: May 11, 2021 at 9:00:00 AM PDT To: "ChimeraX-bugs@cgl.ucsf.edu" <ChimeraX-bugs@cgl.ucsf.edu>
Dear Eric, Thank you so much for your reply.
I have a model of my protein in pdb format, which I am trying to save as .mrc or any sort of volume file on chimera. However, since its a model and do not have any coordinate in the file like xtal or NMR structure, it won’t save it as .mrc or volume file and was giving me the error.
I want to create a volume file from the model to load on cryoSPARC or relion and create 2D projections to help me pick better 2D classes from my raw data. Do you think there is an option to do something like this on chimera?
Thank you again for your time and help in advance – very much appreciated. Sincerely, Roksana
Hi Roksana, No, it only needs an atomic model. You have to open the atomic model first. You don't use molmap with a filename, but instead specify atoms that are already open in ChimeraX. You can use any atom specifier, not necessarily "protein" as in my example. Could be model number, residue numbers, helix, strand, etc. You probably just specified the atoms incorrectly. I could give you a more useful answer if when you ask the question, you included the exact command(s) you tried... then I could see if something was wrong with it. Elaine
On May 11, 2021, at 9:26 AM, Azad, Roksana <razad@gc.cuny.edu> wrote:
Dear Elaine,
I have tried the molmap command in the past, but it shows this error "Missing or invalid "atoms" argument: invalid atoms specifier”. Just to clarify, my protein structure is a model generated from I-tasser, it doesn’t have a resolution. I was thinking that I am getting this error because there are no resolution coordinates in the file?!
Thank you again for your help – very much apppreciated.
Sincerely, Roksana
On May 11, 2021, at 12:09 PM, Elaine Meng <meng@cgl.ucsf.edu> wrote:
Dear Roksana, The command "molmap" creates a map model from an atomic model, for example
open 1zik molmap protein 8
See the "molmap" help for explanation of the resolution value and other command options: <https://urldefense.proofpoint.com/v2/url?u=https-3A__rbvi.ucsf.edu_chimerax_... >
I hope this helps, Elaine ----- Elaine C. Meng, Ph.D. UCSF Chimera(X) team Department of Pharmaceutical Chemistry University of California, San Francisco
Begin forwarded message:
From: "Azad, Roksana" <razad@gc.cuny.edu> Subject: Re: [ChimeraX] #4622: save dialog: 'SaveModelOptionWidget' object has no attribute '_name' Date: May 11, 2021 at 9:00:00 AM PDT To: "ChimeraX-bugs@cgl.ucsf.edu" <ChimeraX-bugs@cgl.ucsf.edu>
Dear Eric, Thank you so much for your reply.
I have a model of my protein in pdb format, which I am trying to save as .mrc or any sort of volume file on chimera. However, since its a model and do not have any coordinate in the file like xtal or NMR structure, it won’t save it as .mrc or volume file and was giving me the error.
I want to create a volume file from the model to load on cryoSPARC or relion and create 2D projections to help me pick better 2D classes from my raw data. Do you think there is an option to do something like this on chimera?
Thank you again for your time and help in advance – very much appreciated. Sincerely, Roksana
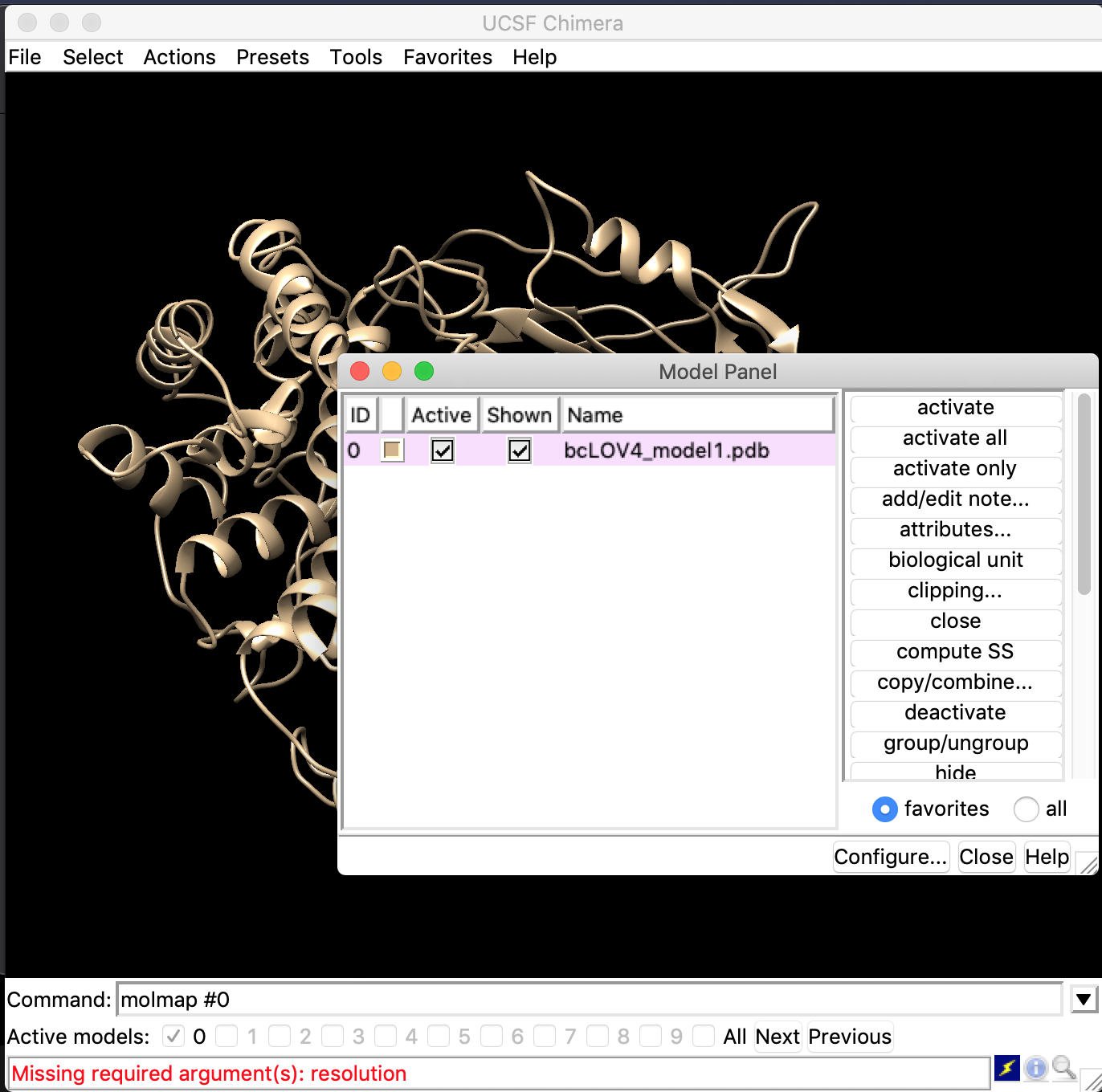
Hi Elaine, I am not sure what you meant, sorry for my ignorance. I attached a screenshot. So, my model number is “0” on chimera when I opened the pdb model, I typed the command “molmap #0” and it says “Missing required argument (s): resolution” (please see the screenshot). I read from the link you sent earlier that I can use “onGrid” options to fix that, but I am a bit confused on how to do that! Many thanks again for your help with this, I appreciate your time. Sincerely, Roksana [cid:3E7BF75B-40F6-4228-A530-FB96DEB8B3B1@home] On May 11, 2021, at 1:33 PM, Elaine Meng <meng@cgl.ucsf.edu<mailto:meng@cgl.ucsf.edu>> wrote: Hi Roksana, No, it only needs an atomic model. You have to open the atomic model first. You don't use molmap with a filename, but instead specify atoms that are already open in ChimeraX. You can use any atom specifier, not necessarily "protein" as in my example. Could be model number, residue numbers, helix, strand, etc. You probably just specified the atoms incorrectly. I could give you a more useful answer if when you ask the question, you included the exact command(s) you tried... then I could see if something was wrong with it. Elaine On May 11, 2021, at 9:26 AM, Azad, Roksana <razad@gc.cuny.edu<mailto:razad@gc.cuny.edu>> wrote: Dear Elaine, I have tried the molmap command in the past, but it shows this error "Missing or invalid "atoms" argument: invalid atoms specifier”. Just to clarify, my protein structure is a model generated from I-tasser, it doesn’t have a resolution. I was thinking that I am getting this error because there are no resolution coordinates in the file?! Thank you again for your help – very much apppreciated. Sincerely, Roksana On May 11, 2021, at 12:09 PM, Elaine Meng <meng@cgl.ucsf.edu<mailto:meng@cgl.ucsf.edu>> wrote: Dear Roksana, The command "molmap" creates a map model from an atomic model, for example open 1zik molmap protein 8 See the "molmap" help for explanation of the resolution value and other command options: <https://urldefense.proofpoint.com/v2/url?u=https-3A__rbvi.ucsf.edu_chimerax_... > I hope this helps, Elaine ----- Elaine C. Meng, Ph.D. UCSF Chimera(X) team Department of Pharmaceutical Chemistry University of California, San Francisco Begin forwarded message: From: "Azad, Roksana" <razad@gc.cuny.edu<mailto:razad@gc.cuny.edu>> Subject: Re: [ChimeraX] #4622: save dialog: 'SaveModelOptionWidget' object has no attribute '_name' Date: May 11, 2021 at 9:00:00 AM PDT To: "ChimeraX-bugs@cgl.ucsf.edu<mailto:ChimeraX-bugs@cgl.ucsf.edu>" <ChimeraX-bugs@cgl.ucsf.edu<mailto:ChimeraX-bugs@cgl.ucsf.edu>> Dear Eric, Thank you so much for your reply. I have a model of my protein in pdb format, which I am trying to save as .mrc or any sort of volume file on chimera. However, since its a model and do not have any coordinate in the file like xtal or NMR structure, it won’t save it as .mrc or volume file and was giving me the error. I want to create a volume file from the model to load on cryoSPARC or relion and create 2D projections to help me pick better 2D classes from my raw data. Do you think there is an option to do something like this on chimera? Thank you again for your time and help in advance – very much appreciated. Sincerely, Roksana
{kind=link}
You sent your message to ChimeraX. Now I see you are using Chimera. So first, you have to tell us which program you are using!!!! ChimeraX questions should go to chimerax-users@cgl.ucsf.edu and Chimera questions should go to chimera-users@cgl.ucsf.edu However, in this case the answer is the same for both programs. You should look at the help page for the "molmap" command of whichever program you are actually using. It says that after the atoms you have to give a number in the command. My example was "molmap protein 8" but you could use "molmap #0 8" or "molmap #0 4.5" etc. Please look at the help page to see the explanation of the number, the resolution parameter, so you can decide what value is appropriate to use. In ChimeraX, there is no #0 (models start with #1) but it also requires the resolution value. Elaine
On May 11, 2021, at 10:46 AM, Azad, Roksana <razad@gc.cuny.edu> wrote:
Hi Elaine,
I am not sure what you meant, sorry for my ignorance. I attached a screenshot. So, my model number is “0” on chimera when I opened the pdb model, I typed the command “molmap #0” and it says “Missing required argument (s): resolution” (please see the screenshot). I read from the link you sent earlier that I can use “onGrid” options to fix that, but I am a bit confused on how to do that! Many thanks again for your help with this, I appreciate your time.
Sincerely, Roksana <RA_ScreenCapture 2021-05-11 at 1.37.45 PM.png>
On May 11, 2021, at 1:33 PM, Elaine Meng <meng@cgl.ucsf.edu> wrote:
Hi Roksana, No, it only needs an atomic model. You have to open the atomic model first. You don't use molmap with a filename, but instead specify atoms that are already open in ChimeraX. You can use any atom specifier, not necessarily "protein" as in my example. Could be model number, residue numbers, helix, strand, etc.
You probably just specified the atoms incorrectly. I could give you a more useful answer if when you ask the question, you included the exact command(s) you tried... then I could see if something was wrong with it. Elaine
On May 11, 2021, at 9:26 AM, Azad, Roksana <razad@gc.cuny.edu> wrote:
Dear Elaine,
I have tried the molmap command in the past, but it shows this error "Missing or invalid "atoms" argument: invalid atoms specifier”. Just to clarify, my protein structure is a model generated from I-tasser, it doesn’t have a resolution. I was thinking that I am getting this error because there are no resolution coordinates in the file?!
Thank you again for your help – very much apppreciated.
Sincerely, Roksana
On May 11, 2021, at 12:09 PM, Elaine Meng <meng@cgl.ucsf.edu> wrote:
Dear Roksana, The command "molmap" creates a map model from an atomic model, for example
open 1zik molmap protein 8
See the "molmap" help for explanation of the resolution value and other command options: <https://urldefense.proofpoint.com/v2/url?u=https-3A__rbvi.ucsf.edu_chimerax_... >
I hope this helps, Elaine ----- Elaine C. Meng, Ph.D. UCSF Chimera(X) team Department of Pharmaceutical Chemistry University of California, San Francisco
Begin forwarded message:
From: "Azad, Roksana" <razad@gc.cuny.edu> Subject: Re: [ChimeraX] #4622: save dialog: 'SaveModelOptionWidget' object has no attribute '_name' Date: May 11, 2021 at 9:00:00 AM PDT To: "ChimeraX-bugs@cgl.ucsf.edu" <ChimeraX-bugs@cgl.ucsf.edu>
Dear Eric, Thank you so much for your reply.
I have a model of my protein in pdb format, which I am trying to save as .mrc or any sort of volume file on chimera. However, since its a model and do not have any coordinate in the file like xtal or NMR structure, it won’t save it as .mrc or volume file and was giving me the error.
I want to create a volume file from the model to load on cryoSPARC or relion and create 2D projections to help me pick better 2D classes from my raw data. Do you think there is an option to do something like this on chimera?
Thank you again for your time and help in advance – very much appreciated. Sincerely, Roksana
Hi Elaine, It worked! I only had to set up resolution (8 or 10) after the molmap command!!!! Thank you so much, Roksana
On May 11, 2021, at 1:57 PM, Elaine Meng <meng@cgl.ucsf.edu> wrote:
You sent your message to ChimeraX. Now I see you are using Chimera. So first, you have to tell us which program you are using!!!!
ChimeraX questions should go to chimerax-users@cgl.ucsf.edu and Chimera questions should go to chimera-users@cgl.ucsf.edu
However, in this case the answer is the same for both programs. You should look at the help page for the "molmap" command of whichever program you are actually using. It says that after the atoms you have to give a number in the command. My example was "molmap protein 8" but you could use "molmap #0 8" or "molmap #0 4.5" etc. Please look at the help page to see the explanation of the number, the resolution parameter, so you can decide what value is appropriate to use. In ChimeraX, there is no #0 (models start with #1) but it also requires the resolution value.
Elaine
On May 11, 2021, at 10:46 AM, Azad, Roksana <razad@gc.cuny.edu> wrote:
Hi Elaine,
I am not sure what you meant, sorry for my ignorance. I attached a screenshot. So, my model number is “0” on chimera when I opened the pdb model, I typed the command “molmap #0” and it says “Missing required argument (s): resolution” (please see the screenshot). I read from the link you sent earlier that I can use “onGrid” options to fix that, but I am a bit confused on how to do that! Many thanks again for your help with this, I appreciate your time.
Sincerely, Roksana <RA_ScreenCapture 2021-05-11 at 1.37.45 PM.png>
On May 11, 2021, at 1:33 PM, Elaine Meng <meng@cgl.ucsf.edu> wrote:
Hi Roksana, No, it only needs an atomic model. You have to open the atomic model first. You don't use molmap with a filename, but instead specify atoms that are already open in ChimeraX. You can use any atom specifier, not necessarily "protein" as in my example. Could be model number, residue numbers, helix, strand, etc.
You probably just specified the atoms incorrectly. I could give you a more useful answer if when you ask the question, you included the exact command(s) you tried... then I could see if something was wrong with it. Elaine
On May 11, 2021, at 9:26 AM, Azad, Roksana <razad@gc.cuny.edu> wrote:
Dear Elaine,
I have tried the molmap command in the past, but it shows this error "Missing or invalid "atoms" argument: invalid atoms specifier”. Just to clarify, my protein structure is a model generated from I-tasser, it doesn’t have a resolution. I was thinking that I am getting this error because there are no resolution coordinates in the file?!
Thank you again for your help – very much apppreciated.
Sincerely, Roksana
On May 11, 2021, at 12:09 PM, Elaine Meng <meng@cgl.ucsf.edu> wrote:
Dear Roksana, The command "molmap" creates a map model from an atomic model, for example
open 1zik molmap protein 8
See the "molmap" help for explanation of the resolution value and other command options: <https://urldefense.proofpoint.com/v2/url?u=https-3A__rbvi.ucsf.edu_chimerax_... >
I hope this helps, Elaine ----- Elaine C. Meng, Ph.D. UCSF Chimera(X) team Department of Pharmaceutical Chemistry University of California, San Francisco
Begin forwarded message:
From: "Azad, Roksana" <razad@gc.cuny.edu> Subject: Re: [ChimeraX] #4622: save dialog: 'SaveModelOptionWidget' object has no attribute '_name' Date: May 11, 2021 at 9:00:00 AM PDT To: "ChimeraX-bugs@cgl.ucsf.edu" <ChimeraX-bugs@cgl.ucsf.edu>
Dear Eric, Thank you so much for your reply.
I have a model of my protein in pdb format, which I am trying to save as .mrc or any sort of volume file on chimera. However, since its a model and do not have any coordinate in the file like xtal or NMR structure, it won’t save it as .mrc or volume file and was giving me the error.
I want to create a volume file from the model to load on cryoSPARC or relion and create 2D projections to help me pick better 2D classes from my raw data. Do you think there is an option to do something like this on chimera?
Thank you again for your time and help in advance – very much appreciated. Sincerely, Roksana
participants (3)
-
 Azad, Roksana
Azad, Roksana -
 Elaine Meng
Elaine Meng -
 Eric Pettersen
Eric Pettersen