Boltz run- Kd values

Hello, I am a Research Scientist at Mount Sinai School of Medicine, NY. I have run Boltz in ChimeraX on a protein-ligand complex, however, I am not able to locate the file for the predicted Kd values. Can you please direct me to the file/location? I am attaching a screenshot of what I see after the run. Thanks a lot, Radhika ________________________________ Confidentiality Notice: This electronic mail transmission and any documents, files, or previous email messages attached to it may contain confidential information and are intended solely for the use of the individual or entity to which they are addressed. If you are not the intended recipient, or a person responsible for delivering such information to the intended recipient, you are hereby notified that any further review, disclosure, copying, dissemination, distribution, or use of any of the information contained in or attached to this email transmission is strictly prohibited. If you have received this message in error, please notify the sender immediately by email, discard any paper copies, and delete all electronic files of the message. Thank you for your cooperation.
{kind=link}

Hi Radhika, Maybe you are using an older version. The current version of the Boltz tool (in ChimeraX 1.11 since July 22, 2025) has an option "Predict ligand binding affinity..." and I do not see that in your image, so maybe there isn't any such prediction in your results. See current help pages: <https://rbvi.ucsf.edu/chimerax/docs/user/tools/boltz.html#dialog> <https://www.rbvi.ucsf.edu/chimerax/data/boltz-apr2025/boltz_help.html> So you may need to get ChimeraX 1.11 and rerun your calculation with the option turned on to get the affinity prediction. Regards, Elaine ----- Elaine C. Meng, Ph.D. UCSF Chimera(X) team Department of Pharmaceutical Chemistry University of California, San Francisco
On Dec 4, 2025, at 10:54 AM, Malik, Radhika via ChimeraX-users <chimerax-users@cgl.ucsf.edu> wrote:
Hello,
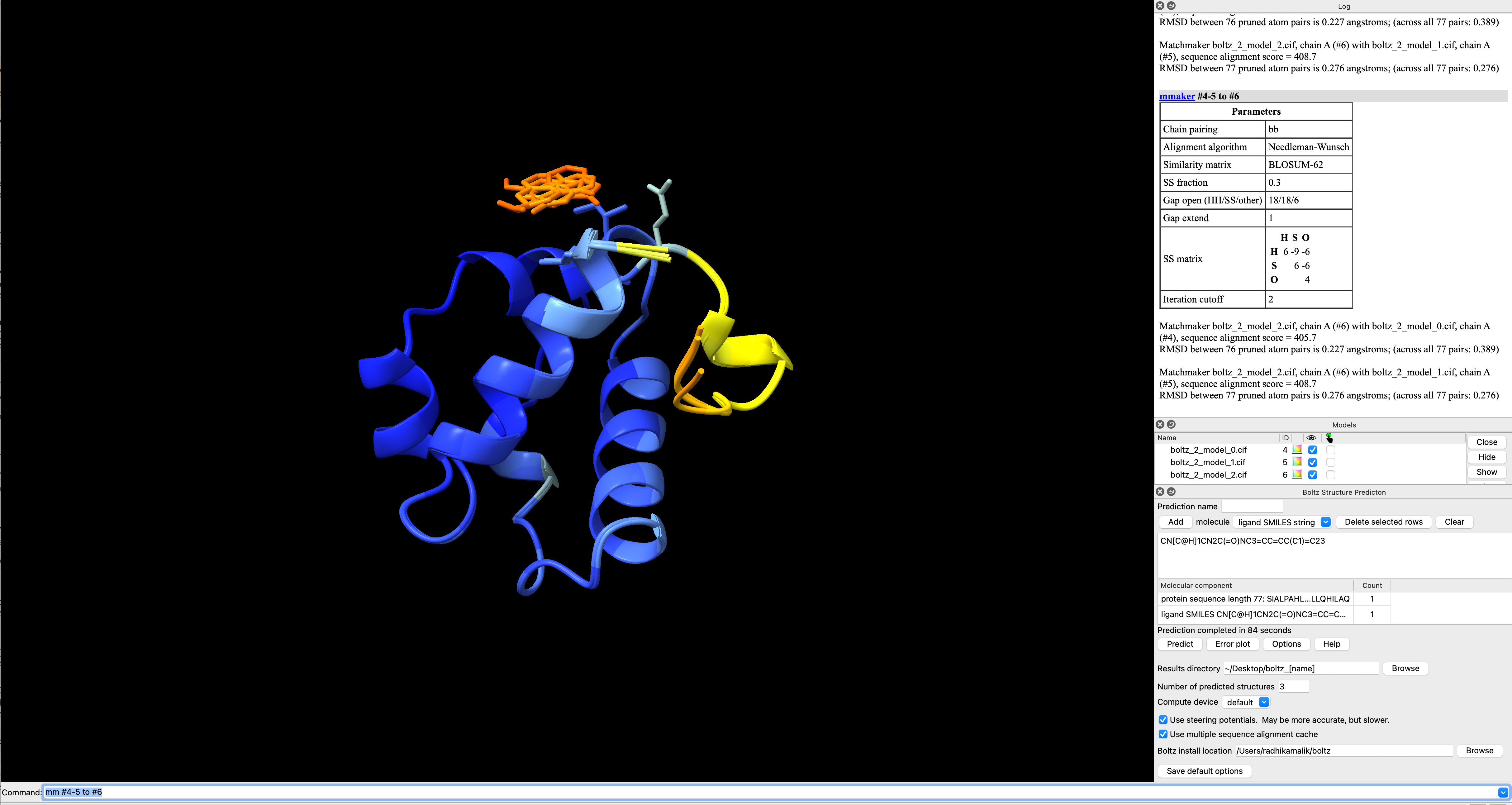
I am a Research Scientist at Mount Sinai School of Medicine, NY. I have run Boltz in ChimeraX on a protein-ligand complex, however, I am not able to locate the file for the predicted Kd values. Can you please direct me to the file/location? I am attaching a screenshot of what I see after the run.
Thanks a lot, Radhika <Screenshot 2025-12-04 at 1.52.02 PM.png>

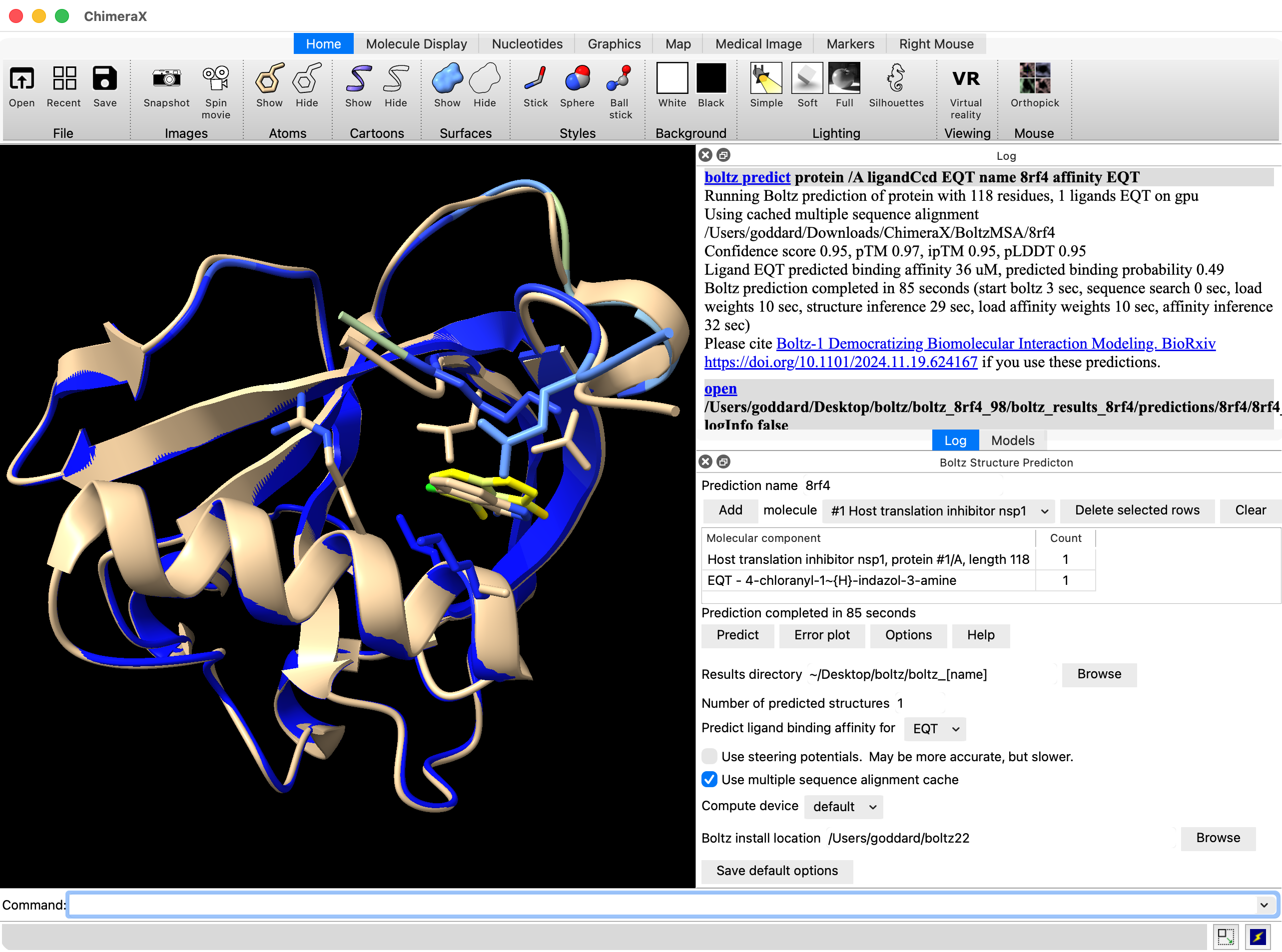
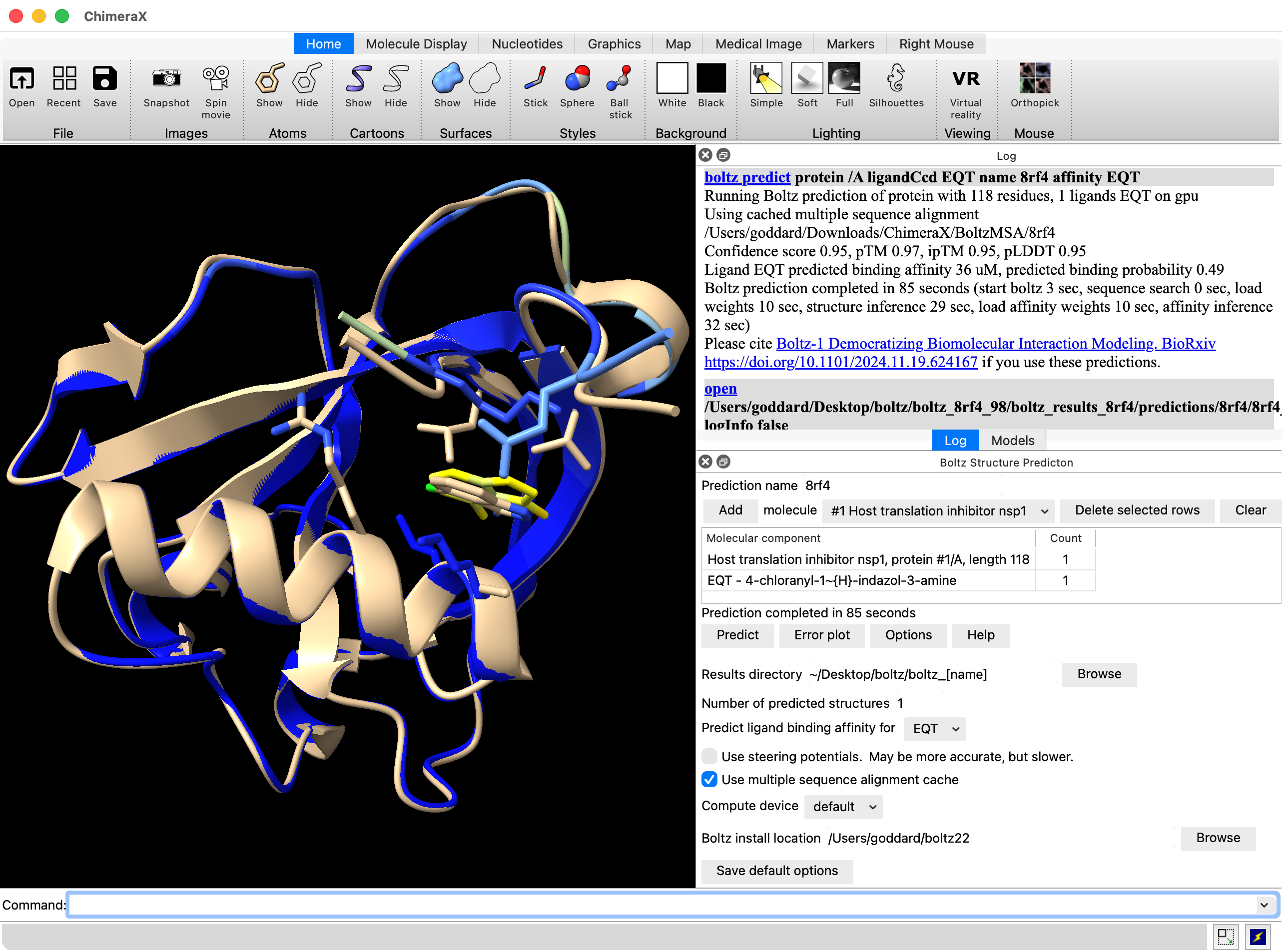
Hi Radhika, Elaine is right. You are probably using ChimeraX 1.10 from June 2025 and Boltz ligand affinity prediction came out after that release. So you need to get a current ChimeraX release candidate or daily build to predict affinity. Also the affinity should not be treated as a real Kd value, it is just a qualitative score. Here is what a prediction with ligand affinity looks like in the ChimeraX daily build. The log reports the affinity as 36 uM and also has the "open" command which shows where the data is located on your computer, in this example, open /Users/goddard/Desktop/boltz/boltz_8rf4_98/boltz_results_8rf4/predictions/8rf4/8rf4_model_0.cif A text file with confidence scores will be found in the same directory. Tom
On Dec 4, 2025, at 12:51 PM, Elaine Meng via ChimeraX-users <chimerax-users@cgl.ucsf.edu> wrote:
Hi Radhika, Maybe you are using an older version. The current version of the Boltz tool (in ChimeraX 1.11 since July 22, 2025) has an option "Predict ligand binding affinity..." and I do not see that in your image, so maybe there isn't any such prediction in your results. See current help pages:
<https://rbvi.ucsf.edu/chimerax/docs/user/tools/boltz.html#dialog> <https://www.rbvi.ucsf.edu/chimerax/data/boltz-apr2025/boltz_help.html>
So you may need to get ChimeraX 1.11 and rerun your calculation with the option turned on to get the affinity prediction.
Regards, Elaine ----- Elaine C. Meng, Ph.D. UCSF Chimera(X) team Department of Pharmaceutical Chemistry University of California, San Francisco
On Dec 4, 2025, at 10:54 AM, Malik, Radhika via ChimeraX-users <chimerax-users@cgl.ucsf.edu> wrote:
Hello,
I am a Research Scientist at Mount Sinai School of Medicine, NY. I have run Boltz in ChimeraX on a protein-ligand complex, however, I am not able to locate the file for the predicted Kd values. Can you please direct me to the file/location? I am attaching a screenshot of what I see after the run.
Thanks a lot, Radhika <Screenshot 2025-12-04 at 1.52.02 PM.png>
_______________________________________________ ChimeraX-users mailing list -- chimerax-users@cgl.ucsf.edu To unsubscribe send an email to chimerax-users-leave@cgl.ucsf.edu Archives: https://mail.cgl.ucsf.edu/mailman/archives/list/chimerax-users@cgl.ucsf.edu/
{kind=link}
Thanks Tom and Elaine! Best, Radhika Sent from my iPhone On Dec 4, 2025, at 4:34 PM, Tom Goddard <goddard@sonic.net> wrote: USE CAUTION: External Message. Hi Radhika, Elaine is right. You are probably using ChimeraX 1.10 from June 2025 and Boltz ligand affinity prediction came out after that release. So you need to get a current ChimeraX release candidate or daily build to predict affinity. Also the affinity should not be treated as a real Kd value, it is just a qualitative score. Here is what a prediction with ligand affinity looks like in the ChimeraX daily build. The log reports the affinity as 36 uM and also has the "open" command which shows where the data is located on your computer, in this example, open /Users/goddard/Desktop/boltz/boltz_8rf4_98/boltz_results_8rf4/predictions/8rf4/8rf4_model_0.cif A text file with confidence scores will be found in the same directory. Tom <boltz_affinity.png>
On Dec 4, 2025, at 12:51 PM, Elaine Meng via ChimeraX-users <chimerax-users@cgl.ucsf.edu> wrote:
Hi Radhika, Maybe you are using an older version. The current version of the Boltz tool (in ChimeraX 1.11 since July 22, 2025) has an option "Predict ligand binding affinity..." and I do not see that in your image, so maybe there isn't any such prediction in your results. See current help pages:
<https://urldefense.proofpoint.com/v2/url?u=https-3A__rbvi.ucsf.edu_chimerax_... > <https://urldefense.proofpoint.com/v2/url?u=https-3A__www.rbvi.ucsf.edu_chime... >
So you may need to get ChimeraX 1.11 and rerun your calculation with the option turned on to get the affinity prediction.
Regards, Elaine ----- Elaine C. Meng, Ph.D. UCSF Chimera(X) team Department of Pharmaceutical Chemistry University of California, San Francisco
On Dec 4, 2025, at 10:54 AM, Malik, Radhika via ChimeraX-users <chimerax-users@cgl.ucsf.edu> wrote:
Hello,
I am a Research Scientist at Mount Sinai School of Medicine, NY. I have run Boltz in ChimeraX on a protein-ligand complex, however, I am not able to locate the file for the predicted Kd values. Can you please direct me to the file/location? I am attaching a screenshot of what I see after the run.
Thanks a lot, Radhika <Screenshot 2025-12-04 at 1.52.02 PM.png>
_______________________________________________ ChimeraX-users mailing list -- chimerax-users@cgl.ucsf.edu To unsubscribe send an email to chimerax-users-leave@cgl.ucsf.edu Archives: https://urldefense.proofpoint.com/v2/url?u=https-3A__mail.cgl.ucsf.edu_mailm...
________________________________ Confidentiality Notice: This electronic mail transmission and any documents, files, or previous email messages attached to it may contain confidential information and are intended solely for the use of the individual or entity to which they are addressed. If you are not the intended recipient, or a person responsible for delivering such information to the intended recipient, you are hereby notified that any further review, disclosure, copying, dissemination, distribution, or use of any of the information contained in or attached to this email transmission is strictly prohibited. If you have received this message in error, please notify the sender immediately by email, discard any paper copies, and delete all electronic files of the message. Thank you for your cooperation.
{kind=link}

Hi all, does Boltz allows to define the binding region? Thanks for your help, Best regards, Marco Sette -- Marco Sette, PhD Associate Professor of Molecular Biology Department of Chemical Sciences and Technology The University of Rome, "Tor Vergata" Via della Ricerca Scientifica, 00133, Rome, Italy e-mail: sette@uniroma2.it Tel.: +39-0672594424 Fax: +39-0672594328
Hi Marco, Boltz allows specifying where a ligand should bind. It is called a "pocket constraint" where you can specify residue numbers in the protein that the ligand should be near. ChimeraX doesn't have a user interface to set this. But you could still run a ChimeraX prediction without a pocket constraint, then edit the Boltz .yaml input text file produced by ChimeraX to add the pocket constraint, and rerun Boltz from a terminal. I described how to do a similar rerunning to add modified protein residues in this mailing list message https://mail.cgl.ucsf.edu/mailman/archives/list/chimerax-users@cgl.ucsf.edu/... The Boltz documentation for specifying a pocket constraint is here https://github.com/jwohlwend/boltz/blob/main/docs/prediction.md I plan to add ChimeraX user interfaces to do these fancier Boltz predictions such as pocket restraints, atom-atom distance restraints, covalently bound ligands, and post-translational modifications, but I haven't done it yet. There is a reason I have been a bit slow to add those features. When I have used those capabilities when running Boltz by hand they very often gave bad results. For example, I had a ligand binding prediction that put the ligand in the wrong place. So I added a pocket constraint and then it did put the ligand in the right pocket. But the pose and the binding affinity were all wrong -- I knew because I had an experimental structure. My suspicion is that if you have to tell Boltz to put the ligand somewhere where it doesn't want to put it, the chance of getting a good pose or affinity prediction is small. There is a reason why it didn't put the ligand there without the constraint -- it doesn't think the ligand goes there, and forcing it only has a small chance of allowing Boltz to figure out the right binding pose. Tom
On Dec 5, 2025, at 1:30 AM, Marco Sette via ChimeraX-users <chimerax-users@cgl.ucsf.edu> wrote:
Hi all,
does Boltz allows to define the binding region?
Thanks for your help,
Best regards,
Marco Sette
-- Marco Sette, PhD Associate Professor of Molecular Biology Department of Chemical Sciences and Technology The University of Rome, "Tor Vergata" Via della Ricerca Scientifica, 00133, Rome, Italy e-mail: sette@uniroma2.it Tel.: +39-0672594424 Fax: +39-0672594328
_______________________________________________ ChimeraX-users mailing list -- chimerax-users@cgl.ucsf.edu To unsubscribe send an email to chimerax-users-leave@cgl.ucsf.edu Archives: https://mail.cgl.ucsf.edu/mailman/archives/list/chimerax-users@cgl.ucsf.edu/
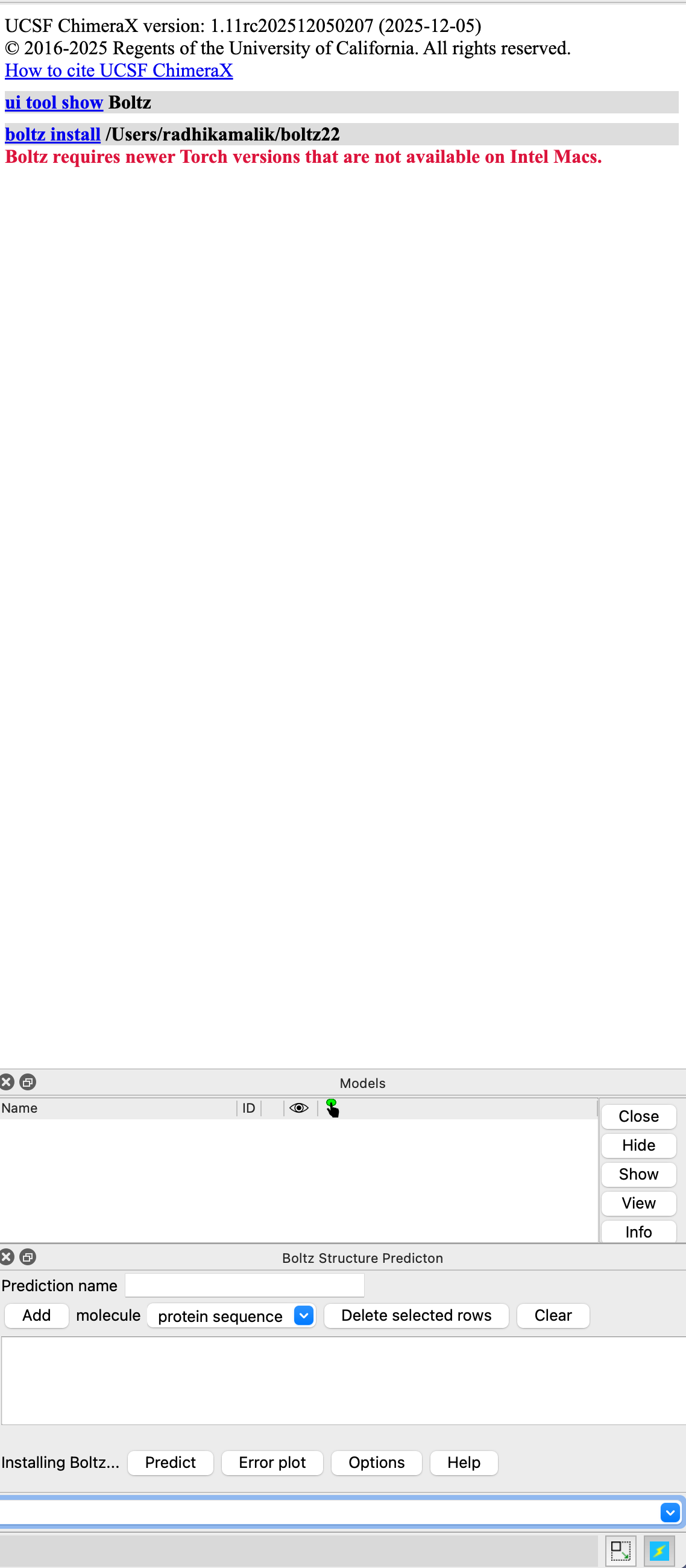
Hi Tom and Elaine, I am trying to install boltz after installing chimerax 1.11 but it seems to be stalling, there is no error as such (see attached). I have a apple M4 max and was able to run boltz on chimerax 1.10 fine. Thanks a lot, Radhika ________________________________ From: Malik, Radhika Sent: Friday, December 5, 2025 9:45 AM To: Tom Goddard <goddard@sonic.net> Cc: Mailing List <chimerax-users@cgl.ucsf.edu> Subject: Re: [chimerax-users] Boltz run- Kd values Thanks Tom and Elaine! Best, Radhika Sent from my iPhone On Dec 4, 2025, at 4:34 PM, Tom Goddard <goddard@sonic.net> wrote: USE CAUTION: External Message. Hi Radhika, Elaine is right. You are probably using ChimeraX 1.10 from June 2025 and Boltz ligand affinity prediction came out after that release. So you need to get a current ChimeraX release candidate or daily build to predict affinity. Also the affinity should not be treated as a real Kd value, it is just a qualitative score. Here is what a prediction with ligand affinity looks like in the ChimeraX daily build. The log reports the affinity as 36 uM and also has the "open" command which shows where the data is located on your computer, in this example, open /Users/goddard/Desktop/boltz/boltz_8rf4_98/boltz_results_8rf4/predictions/8rf4/8rf4_model_0.cif A text file with confidence scores will be found in the same directory. Tom <boltz_affinity.png>
On Dec 4, 2025, at 12:51 PM, Elaine Meng via ChimeraX-users <chimerax-users@cgl.ucsf.edu> wrote:
Hi Radhika, Maybe you are using an older version. The current version of the Boltz tool (in ChimeraX 1.11 since July 22, 2025) has an option "Predict ligand binding affinity..." and I do not see that in your image, so maybe there isn't any such prediction in your results. See current help pages:
<https://urldefense.proofpoint.com/v2/url?u=https-3A__rbvi.ucsf.edu_chimerax_... > <https://urldefense.proofpoint.com/v2/url?u=https-3A__www.rbvi.ucsf.edu_chime... >
So you may need to get ChimeraX 1.11 and rerun your calculation with the option turned on to get the affinity prediction.
Regards, Elaine ----- Elaine C. Meng, Ph.D. UCSF Chimera(X) team Department of Pharmaceutical Chemistry University of California, San Francisco
On Dec 4, 2025, at 10:54 AM, Malik, Radhika via ChimeraX-users <chimerax-users@cgl.ucsf.edu> wrote:
Hello,
I am a Research Scientist at Mount Sinai School of Medicine, NY. I have run Boltz in ChimeraX on a protein-ligand complex, however, I am not able to locate the file for the predicted Kd values. Can you please direct me to the file/location? I am attaching a screenshot of what I see after the run.
Thanks a lot, Radhika <Screenshot 2025-12-04 at 1.52.02 PM.png>
_______________________________________________ ChimeraX-users mailing list -- chimerax-users@cgl.ucsf.edu To unsubscribe send an email to chimerax-users-leave@cgl.ucsf.edu Archives: https://urldefense.proofpoint.com/v2/url?u=https-3A__mail.cgl.ucsf.edu_mailm...
________________________________ Confidentiality Notice: This electronic mail transmission and any documents, files, or previous email messages attached to it may contain confidential information and are intended solely for the use of the individual or entity to which they are addressed. If you are not the intended recipient, or a person responsible for delivering such information to the intended recipient, you are hereby notified that any further review, disclosure, copying, dissemination, distribution, or use of any of the information contained in or attached to this email transmission is strictly prohibited. If you have received this message in error, please notify the sender immediately by email, discard any paper copies, and delete all electronic files of the message. Thank you for your cooperation.
{kind=link}
Hi Radhika, Sorry about that. I have fixed the problem in the ChimeraX daily builds and release candidates that will be made tonight (dated December 5 on download page). I accidentally left some test code in ChimeraX on November 25 to check error messages on old Intel Macs using my ARM Mac, and that got into the daily build and release candidate. It is possible to fix your ChimeraX by editing line 40 of ChimeraX.app/Contents/lib/python3.11/site-packages/chimerax/boltz/install.py from if True or platform.system() == 'Darwin' and platform.machine() == 'x86_64': to if platform.system() == 'Darwin' and platform.machine() == 'x86_64': But I suspect macOS will prevent you from editing this file because it invalidates the application signature, unless you circumvent it using some Mac security setting that allows you to edit applications. Probably best to wait for tomorrow's builds. Tom
On Dec 5, 2025, at 11:31 AM, Malik, Radhika <radhika.malik@mssm.edu> wrote:
Hi Tom and Elaine,
I am trying to install boltz after installing chimerax 1.11 but it seems to be stalling, there is no error as such (see attached). I have a apple M4 max and was able to run boltz on chimerax 1.10 fine.
Thanks a lot, Radhika
From: Malik, Radhika Sent: Friday, December 5, 2025 9:45 AM To: Tom Goddard <goddard@sonic.net> Cc: Mailing List <chimerax-users@cgl.ucsf.edu> Subject: Re: [chimerax-users] Boltz run- Kd values
Thanks Tom and Elaine!
Best, Radhika
Sent from my iPhone
On Dec 4, 2025, at 4:34 PM, Tom Goddard <goddard@sonic.net> wrote:
USE CAUTION: External Message.
Hi Radhika,
Elaine is right. You are probably using ChimeraX 1.10 from June 2025 and Boltz ligand affinity prediction came out after that release. So you need to get a current ChimeraX release candidate or daily build to predict affinity. Also the affinity should not be treated as a real Kd value, it is just a qualitative score.
Here is what a prediction with ligand affinity looks like in the ChimeraX daily build. The log reports the affinity as 36 uM and also has the "open" command which shows where the data is located on your computer, in this example,
open /Users/goddard/Desktop/boltz/boltz_8rf4_98/boltz_results_8rf4/predictions/8rf4/8rf4_model_0.cif
A text file with confidence scores will be found in the same directory.
Tom
<boltz_affinity.png>
On Dec 4, 2025, at 12:51 PM, Elaine Meng via ChimeraX-users <chimerax-users@cgl.ucsf.edu> wrote:
Hi Radhika, Maybe you are using an older version. The current version of the Boltz tool (in ChimeraX 1.11 since July 22, 2025) has an option "Predict ligand binding affinity..." and I do not see that in your image, so maybe there isn't any such prediction in your results. See current help pages:
<https://urldefense.proofpoint.com/v2/url?u=https-3A__rbvi.ucsf.edu_chimerax_... > <https://urldefense.proofpoint.com/v2/url?u=https-3A__www.rbvi.ucsf.edu_chime... >
So you may need to get ChimeraX 1.11 and rerun your calculation with the option turned on to get the affinity prediction.
Regards, Elaine ----- Elaine C. Meng, Ph.D. UCSF Chimera(X) team Department of Pharmaceutical Chemistry University of California, San Francisco
On Dec 4, 2025, at 10:54 AM, Malik, Radhika via ChimeraX-users <chimerax-users@cgl.ucsf.edu> wrote:
Hello,
I am a Research Scientist at Mount Sinai School of Medicine, NY. I have run Boltz in ChimeraX on a protein-ligand complex, however, I am not able to locate the file for the predicted Kd values. Can you please direct me to the file/location? I am attaching a screenshot of what I see after the run.
Thanks a lot, Radhika <Screenshot 2025-12-04 at 1.52.02 PM.png>
_______________________________________________ ChimeraX-users mailing list -- chimerax-users@cgl.ucsf.edu To unsubscribe send an email to chimerax-users-leave@cgl.ucsf.edu Archives: https://urldefense.proofpoint.com/v2/url?u=https-3A__mail.cgl.ucsf.edu_mailm...
Confidentiality Notice: This electronic mail transmission and any documents, files, or previous email messages attached to it may contain confidential information and are intended solely for the use of the individual or entity to which they are addressed. If you are not the intended recipient, or a person responsible for delivering such information to the intended recipient, you are hereby notified that any further review, disclosure, copying, dissemination, distribution, or use of any of the information contained in or attached to this email transmission is strictly prohibited. If you have received this message in error, please notify the sender immediately by email, discard any paper copies, and delete all electronic files of the message. Thank you for your cooperation. <Screenshot 2025-12-05 at 2.27.28 PM.png>
Hi Tom. thanks for the clear explanation. So, it seems that the intelligence behind Boltz is very similar to that of a human and capricious... Thanks again, Marco Il 05/12/2025 19:33, Tom Goddard ha scritto:
Hi Marco,
Boltz allows specifying where a ligand should bind. It is called a "pocket constraint" where you can specify residue numbers in the protein that the ligand should be near. ChimeraX doesn't have a user interface to set this. But you could still run a ChimeraX prediction without a pocket constraint, then edit the Boltz .yaml input text file produced by ChimeraX to add the pocket constraint, and rerun Boltz from a terminal. I described how to do a similar rerunning to add modified protein residues in this mailing list message
https://mail.cgl.ucsf.edu/mailman/archives/list/chimerax-users@cgl.ucsf.edu/... <https://urldefense.com/v3/__https://mail.cgl.ucsf.edu/mailman/archives/list/...>
The Boltz documentation for specifying a pocket constraint is here
https://github.com/jwohlwend/boltz/blob/main/docs/prediction.md <https://urldefense.com/v3/__https://github.com/jwohlwend/boltz/blob/main/doc...>
I plan to add ChimeraX user interfaces to do these fancier Boltz predictions such as pocket restraints, atom-atom distance restraints, covalently bound ligands, and post-translational modifications, but I haven't done it yet. There is a reason I have been a bit slow to add those features. When I have used those capabilities when running Boltz by hand they very often gave bad results. For example, I had a ligand binding prediction that put the ligand in the wrong place. So I added a pocket constraint and then it did put the ligand in the right pocket. But the pose and the binding affinity were all wrong -- I knew because I had an experimental structure. My suspicion is that if you have to tell Boltz to put the ligand somewhere where it doesn't want to put it, the chance of getting a good pose or affinity prediction is small. There is a reason why it didn't put the ligand there without the constraint -- it doesn't think the ligand goes there, and forcing it only has a small chance of allowing Boltz to figure out the right binding pose.
Tom
On Dec 5, 2025, at 1:30 AM, Marco Sette via ChimeraX-users <chimerax-users@cgl.ucsf.edu> wrote:
Hi all,
does Boltz allows to define the binding region?
Thanks for your help,
Best regards,
Marco Sette
-- Marco Sette, PhD Associate Professor of Molecular Biology Department of Chemical Sciences and Technology The University of Rome, "Tor Vergata" Via della Ricerca Scientifica, 00133, Rome, Italy e-mail: sette@uniroma2.it Tel.: +39-0672594424 Fax: +39-0672594328
_______________________________________________ ChimeraX-users mailing list -- chimerax-users@cgl.ucsf.edu To unsubscribe send an email to chimerax-users-leave@cgl.ucsf.edu Archives: https://mail.cgl.ucsf.edu/mailman/archives/list/chimerax-users@cgl.ucsf.edu/ <https://urldefense.com/v3/__https://mail.cgl.ucsf.edu/mailman/archives/list/...>
-- Marco Sette, PhD Associate Professor of Molecular Biology Department of Chemical Sciences and Technology The University of Rome, "Tor Vergata" Via della Ricerca Scientifica, 00133, Rome, Italy e-mail:sette@uniroma2.it Tel.: +39-0672594424 Fax: +39-0672594328 -- Questa email è stata esaminata alla ricerca di virus dal software antivirus Avast. www.avast.com
Thanks Tom! Best, Radhika ________________________________ From: Tom Goddard <goddard@sonic.net> Sent: Friday, December 5, 2025 3:04 PM To: Malik, Radhika <radhika.malik@mssm.edu> Cc: Mailing List <chimerax-users@cgl.ucsf.edu> Subject: Re: [chimerax-users] Boltz run- Kd values Hi Radhika, Sorry about that. I have fixed the problem in the ChimeraX daily builds and release candidates that will be made tonight (dated December 5 on download page). I accidentally left some test code in ChimeraX on November 25 to check error messages on old Intel Macs using my ARM Mac, and that got into the daily build and release candidate. It is possible to fix your ChimeraX by editing line 40 of ChimeraX.app/Contents/lib/python3.11/site-packages/chimerax/boltz/install.py<https://urldefense.proofpoint.com/v2/url?u=http-3A__ChimeraX.app_Contents_li...> from if True or platform.system() == 'Darwin' and platform.machine() == 'x86_64': to if platform.system() == 'Darwin' and platform.machine() == 'x86_64': But I suspect macOS will prevent you from editing this file because it invalidates the application signature, unless you circumvent it using some Mac security setting that allows you to edit applications. Probably best to wait for tomorrow's builds. Tom On Dec 5, 2025, at 11:31 AM, Malik, Radhika <radhika.malik@mssm.edu> wrote: Hi Tom and Elaine, I am trying to install boltz after installing chimerax 1.11 but it seems to be stalling, there is no error as such (see attached). I have a apple M4 max and was able to run boltz on chimerax 1.10 fine. Thanks a lot, Radhika ________________________________ From: Malik, Radhika Sent: Friday, December 5, 2025 9:45 AM To: Tom Goddard <goddard@sonic.net> Cc: Mailing List <chimerax-users@cgl.ucsf.edu> Subject: Re: [chimerax-users] Boltz run- Kd values Thanks Tom and Elaine! Best, Radhika Sent from my iPhone On Dec 4, 2025, at 4:34 PM, Tom Goddard <goddard@sonic.net> wrote: USE CAUTION: External Message. Hi Radhika, Elaine is right. You are probably using ChimeraX 1.10 from June 2025 and Boltz ligand affinity prediction came out after that release. So you need to get a current ChimeraX release candidate or daily build to predict affinity. Also the affinity should not be treated as a real Kd value, it is just a qualitative score. Here is what a prediction with ligand affinity looks like in the ChimeraX daily build. The log reports the affinity as 36 uM and also has the "open" command which shows where the data is located on your computer, in this example, open /Users/goddard/Desktop/boltz/boltz_8rf4_98/boltz_results_8rf4/predictions/8rf4/8rf4_model_0.cif A text file with confidence scores will be found in the same directory. Tom <boltz_affinity.png>
On Dec 4, 2025, at 12:51 PM, Elaine Meng via ChimeraX-users <chimerax-users@cgl.ucsf.edu> wrote:
Hi Radhika, Maybe you are using an older version. The current version of the Boltz tool (in ChimeraX 1.11 since July 22, 2025) has an option "Predict ligand binding affinity..." and I do not see that in your image, so maybe there isn't any such prediction in your results. See current help pages:
<https://urldefense.proofpoint.com/v2/url?u=https-3A__rbvi.ucsf.edu_chimerax_... > <https://urldefense.proofpoint.com/v2/url?u=https-3A__www.rbvi.ucsf.edu_chime... >
So you may need to get ChimeraX 1.11 and rerun your calculation with the option turned on to get the affinity prediction.
Regards, Elaine ----- Elaine C. Meng, Ph.D. UCSF Chimera(X) team Department of Pharmaceutical Chemistry University of California, San Francisco
On Dec 4, 2025, at 10:54 AM, Malik, Radhika via ChimeraX-users <chimerax-users@cgl.ucsf.edu> wrote:
Hello,
I am a Research Scientist at Mount Sinai School of Medicine, NY. I have run Boltz in ChimeraX on a protein-ligand complex, however, I am not able to locate the file for the predicted Kd values. Can you please direct me to the file/location? I am attaching a screenshot of what I see after the run.
Thanks a lot, Radhika <Screenshot 2025-12-04 at 1.52.02 PM.png>
_______________________________________________ ChimeraX-users mailing list -- chimerax-users@cgl.ucsf.edu To unsubscribe send an email to chimerax-users-leave@cgl.ucsf.edu Archives: https://urldefense.proofpoint.com/v2/url?u=https-3A__mail.cgl.ucsf.edu_mailm...
________________________________ Confidentiality Notice: This electronic mail transmission and any documents, files, or previous email messages attached to it may contain confidential information and are intended solely for the use of the individual or entity to which they are addressed. If you are not the intended recipient, or a person responsible for delivering such information to the intended recipient, you are hereby notified that any further review, disclosure, copying, dissemination, distribution, or use of any of the information contained in or attached to this email transmission is strictly prohibited. If you have received this message in error, please notify the sender immediately by email, discard any paper copies, and delete all electronic files of the message. Thank you for your cooperation. <Screenshot 2025-12-05 at 2.27.28 PM.png>
participants (4)
-
 Elaine Meng
Elaine Meng -
 Malik, Radhika
Malik, Radhika -
 Marco Sette
Marco Sette -
 Tom Goddard
Tom Goddard