How to update the bonding status every frame when viewing a molecular dynamic trajectory?

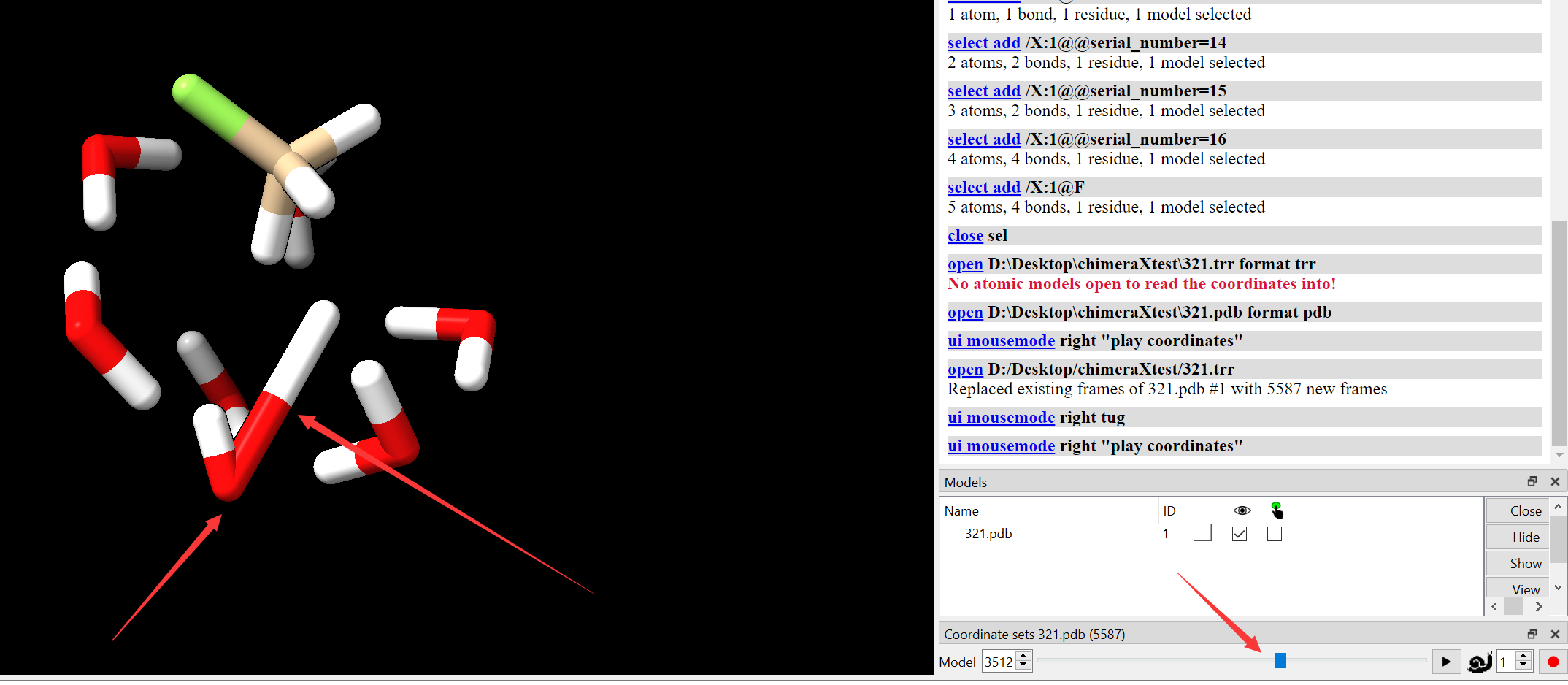
Dear users and developers of ChimeraX, Recently I want to produce some decent movies of a molecular dynamic trajectory, I loaded the pdb file and the trr file into ChimeraX. Everything goes well until some bond broken occurred, the display became very strange since the bond was stretched to an unreasonable length. I have been told a solution by using ~bond, bond #1 to fix the issues. However, it is impossible to do it at every frame when you have to record a movie and your trajectory was with thousands of frames. Thus, I ask for help, is there a solution to update the bonding status of every frame automatically? Yours, Ran
{kind=link}

Dear Ran, You can use the "perframe" command to run something, including a set of other commands, at each graphics frame update. Running ~bond and bond may slow down the live playback, especially if you have lots of atoms, but it won't affect the playback speed of a recorded movie file. It will only be slower when you are doing the live playback and recording. For example, the following ChimeraX commands, using 1plx NMR structure opened as a trajectory: open 1plx coordset true perframe "~bond #1; bond #1" ; coordset #1 1,80; wait 80; ~perframe The "perframe" command says what to do at each frame, and the "coordset" command plays back the trajectory. See the following for details and command options: <https://rbvi.ucsf.edu/chimerax/docs/user/commands/perframe.html> <https://rbvi.ucsf.edu/chimerax/docs/user/commands/coordset.html> Of course you would open your own trajectory instead of 1plx, and make other adjustments as needed for your own data. You could have a "movie record" command before the playback and any other actions you want in your movie, followed by "movie encode" to create the movie file. The movie command also has many options: <https://rbvi.ucsf.edu/chimerax/docs/user/commands/movie.html> See making movies and movie-related commands: <https://rbvi.ucsf.edu/chimerax/docs/user/movies.html> I hope this helps, Elaine ----- Elaine C. Meng, Ph.D. UCSF Chimera(X) team Department of Pharmaceutical Chemistry University of California, San Francisco
On Apr 29, 2023, at 7:23 AM, 洪冉 via ChimeraX-users <chimerax-users@cgl.ucsf.edu> wrote:
Dear users and developers of ChimeraX, Recently I want to produce some decent movies of a molecular dynamic trajectory, I loaded the pdb file and the trr file into ChimeraX. Everything goes well until some bond broken occurred, the display became very strange since the bond was stretched to an unreasonable length. I have been told a solution by using ~bond, bond #1 to fix the issues. However, it is impossible to do it at every frame when you have to record a movie and your trajectory was with thousands of frames. Thus, I ask for help, is there a solution to update the bonding status of every frame automatically?
Yours, Ran <Untitled.png>
Elaine, Thank you very much. It works. Deeply appreciate your detailed and thoughtful guidance. Yours, Ran From: Elaine Meng <meng@cgl.ucsf.edu> Date: 2023-05-01 03:50:19 To: "洪冉" <hongran@ahpu.edu.cn> Cc: chimerax-users@cgl.ucsf.edu Subject: Re: [chimerax-users] How to update the bonding status every frame when viewing a molecular dynamic trajectory?>Dear Ran,
You can use the "perframe" command to run something, including a set of other commands, at each graphics frame update. Running ~bond and bond may slow down the live playback, especially if you have lots of atoms, but it won't affect the playback speed of a recorded movie file. It will only be slower when you are doing the live playback and recording.
For example, the following ChimeraX commands, using 1plx NMR structure opened as a trajectory:
open 1plx coordset true perframe "~bond #1; bond #1" ; coordset #1 1,80; wait 80; ~perframe
The "perframe" command says what to do at each frame, and the "coordset" command plays back the trajectory. See the following for details and command options:
<https://rbvi.ucsf.edu/chimerax/docs/user/commands/perframe.html> <https://rbvi.ucsf.edu/chimerax/docs/user/commands/coordset.html>
Of course you would open your own trajectory instead of 1plx, and make other adjustments as needed for your own data. You could have a "movie record" command before the playback and any other actions you want in your movie, followed by "movie encode" to create the movie file. The movie command also has many options:
<https://rbvi.ucsf.edu/chimerax/docs/user/commands/movie.html>
See making movies and movie-related commands: <https://rbvi.ucsf.edu/chimerax/docs/user/movies.html>
I hope this helps, Elaine ----- Elaine C. Meng, Ph.D. UCSF Chimera(X) team Department of Pharmaceutical Chemistry University of California, San Francisco
On Apr 29, 2023, at 7:23 AM, 洪冉 via ChimeraX-users <chimerax-users@cgl.ucsf.edu> wrote:
Dear users and developers of ChimeraX, Recently I want to produce some decent movies of a molecular dynamic trajectory, I loaded the pdb file and the trr file into ChimeraX. Everything goes well until some bond broken occurred, the display became very strange since the bond was stretched to an unreasonable length. I have been told a solution by using ~bond, bond #1 to fix the issues. However, it is impossible to do it at every frame when you have to record a movie and your trajectory was with thousands of frames. Thus, I ask for help, is there a solution to update the bonding status of every frame automatically?
Yours, Ran <Untitled.png>
participants (2)
-
 Elaine Meng
Elaine Meng -
 洪冉
洪冉