
Hello Chimera/ChimeraX team I recently asked already a question regarding the saving/exporting of vertices generated during the surface calculation in ChimeraX and the associated parameters with this surface generation. On a related note, I wanted to ask whether it is possible to also extract the surface (SESA and/or SASA) on a per atom basis. MSMS calculates not only the vertices for each atom (usually several vertices per atom) but it also calculates the SESA and SASA on a per atom basis. Now, I was wondering whether something is possible in ChimeraX too. As I was parsing through the docs, I was wondering whether ChimeraX calculates the surface no longer using MSMS. The vertices density supported by ChimeraX is considerably higher than what I can get with my standalone MSMS program (at least factor 6). So, if ChimeraX does not longer employ MSMS, what else does it use? With "MEASURE CONVEXITY" I get the surface vertex positions, normals, convexity values, and triangles. Unfortunately, the association with the corresponding residue is lost (however, can be regained with a little bit of work I guess, simply using euclidean distance). Question: Is there something similar for the SESA or SASA of the different atoms in the model/chain. In theory, this information should be available, at least the SESA. I am just wondering whether there is a way to write this out as a raw txt file in ChimeraX The closest I found in the save command is the .obj or .vtk format. However, both are semi-satisfying in that that they both do not provide the data looked for and are in somewhat special formats, at least the vtk. The .obj file is much much bigger than what MSMS or "measure convexity" generates, and not really handy for then working with the data in other programs, mainly python. As always, I very much appreciate any help and wish to thank the team for their contributions to the field. Best regards Dennis

Hi Dennis, If you use the "measure sasa" command in ChimeraX, you can assign SASA values per atom and residue as attributes named "area": <https://rbvi.ucsf.edu/chimerax/docs/user/commands/measure.html#sasa> Attributes can be used for specification, coloring, etc., and written to a file with "save" <https://rbvi.ucsf.edu/chimerax/docs/user/attributes.html> <https://rbvi.ucsf.edu/chimerax/docs/user/commands/save.html#attributes> SESA is not available, however. ChimeraX does use a different method than Chimera to calculate the molecular surface, much more robust (less prone to failures). <https://rbvi.ucsf.edu/chimerax/docs/user/commands/surface.html> I hope this helps, Elaine ----- Elaine C. Meng, Ph.D. UCSF Chimera(X) team Department of Pharmaceutical Chemistry University of California, San Francisco
On Oct 19, 2023, at 4:33 AM, Dennis Dannecker via ChimeraX-users <chimerax-users@cgl.ucsf.edu> wrote:
Hello Chimera/ChimeraX team
I recently asked already a question regarding the saving/exporting of vertices generated during the surface calculation in ChimeraX and the associated parameters with this surface generation.
On a related note, I wanted to ask whether it is possible to also extract the surface (SESA and/or SASA) on a per atom basis. MSMS calculates not only the vertices for each atom (usually several vertices per atom) but it also calculates the SESA and SASA on a per atom basis. Now, I was wondering whether something is possible in ChimeraX too. As I was parsing through the docs, I was wondering whether ChimeraX calculates the surface no longer using MSMS. The vertices density supported by ChimeraX is considerably higher than what I can get with my standalone MSMS program (at least factor 6). So, if ChimeraX does not longer employ MSMS, what else does it use?
With "measure convexity" I get the surface vertex positions, normals, convexity values, and triangles. Unfortunately, the association with the corresponding residue is lost (however, can be regained with a little bit of work I guess, simply using euclidean distance).
Question: Is there something similar for the SESA or SASA of the different atoms in the model/chain. In theory, this information should be available, at least the SESA. I am just wondering whether there is a way to write this out as a raw txt file in ChimeraX
The closest I found in the save command is the .obj or .vtk format. However, both are semi-satisfying in that that they both do not provide the data looked for and are in somewhat special formats, at least the vtk. The .obj file is much much bigger than what MSMS or "measure convexity" generates, and not really handy for then working with the data in other programs, mainly python.
As always, I very much appreciate any help and wish to thank the team for their contributions to the field.
Best regards
Dennis

ChimeraX knows which atom every surface vertex is associated with. But if you just want the solvent accessible surface area per atom then use the "measure sasa" command as Elaine suggested. The per-atom area calculation does not use the displayed triangles. Instead it does an analytic calculation that is described here https://www.cgl.ucsf.edu/chimera/data/sasa-nov2013/sasa.html The MSMS solvent excluded surface calculation in Chimera failed on many large atomic models (> 10,000 atoms) so we replaced it with a grid based solvent excluded surface algorithm which reliably computes surfaces for any size atomic model. Tom
On Oct 19, 2023, at 9:55 AM, Elaine Meng via ChimeraX-users <chimerax-users@cgl.ucsf.edu> wrote:
Hi Dennis, If you use the "measure sasa" command in ChimeraX, you can assign SASA values per atom and residue as attributes named "area":
<https://rbvi.ucsf.edu/chimerax/docs/user/commands/measure.html#sasa>
Attributes can be used for specification, coloring, etc., and written to a file with "save"
<https://rbvi.ucsf.edu/chimerax/docs/user/attributes.html> <https://rbvi.ucsf.edu/chimerax/docs/user/commands/save.html#attributes>
SESA is not available, however. ChimeraX does use a different method than Chimera to calculate the molecular surface, much more robust (less prone to failures).
<https://rbvi.ucsf.edu/chimerax/docs/user/commands/surface.html>
I hope this helps, Elaine ----- Elaine C. Meng, Ph.D. UCSF Chimera(X) team Department of Pharmaceutical Chemistry University of California, San Francisco
On Oct 19, 2023, at 4:33 AM, Dennis Dannecker via ChimeraX-users <chimerax-users@cgl.ucsf.edu> wrote:
Hello Chimera/ChimeraX team
I recently asked already a question regarding the saving/exporting of vertices generated during the surface calculation in ChimeraX and the associated parameters with this surface generation.
On a related note, I wanted to ask whether it is possible to also extract the surface (SESA and/or SASA) on a per atom basis. MSMS calculates not only the vertices for each atom (usually several vertices per atom) but it also calculates the SESA and SASA on a per atom basis. Now, I was wondering whether something is possible in ChimeraX too. As I was parsing through the docs, I was wondering whether ChimeraX calculates the surface no longer using MSMS. The vertices density supported by ChimeraX is considerably higher than what I can get with my standalone MSMS program (at least factor 6). So, if ChimeraX does not longer employ MSMS, what else does it use?
With "measure convexity" I get the surface vertex positions, normals, convexity values, and triangles. Unfortunately, the association with the corresponding residue is lost (however, can be regained with a little bit of work I guess, simply using euclidean distance).
Question: Is there something similar for the SESA or SASA of the different atoms in the model/chain. In theory, this information should be available, at least the SESA. I am just wondering whether there is a way to write this out as a raw txt file in ChimeraX
The closest I found in the save command is the .obj or .vtk format. However, both are semi-satisfying in that that they both do not provide the data looked for and are in somewhat special formats, at least the vtk. The .obj file is much much bigger than what MSMS or "measure convexity" generates, and not really handy for then working with the data in other programs, mainly python.
As always, I very much appreciate any help and wish to thank the team for their contributions to the field.
Best regards
Dennis
_______________________________________________ ChimeraX-users mailing list -- chimerax-users@cgl.ucsf.edu To unsubscribe send an email to chimerax-users-leave@cgl.ucsf.edu Archives: https://mail.cgl.ucsf.edu/mailman/archives/list/chimerax-users@cgl.ucsf.edu/
Hello Tom, thank you very much for your reply. Since you were saying, "ChimeraX knows which atom every surface vertex is associated with." , as a matter of fact, I would like to have this vertex-atom assignment. If there is an easy way to write this out as a file, then I would definitely go with that. As of now, I do the assignment with simply finding the closest atom for each vertex with a small script, it is reasonably fast thanks to vectorization and multiprocessing. But more elegant of cours ewould be to use what ChimeraX has already calculated. I have realized that the calculated surface area per atom is not affected by the gridSpacing parameter used for surface calculation. Now that I know that ChimeraX uses an analytic calculation that independence makes of course sense. Having read your work on the SASA from Nov 2013 (https://www.cgl.ucsf.edu/chimera/data/sasa-nov2013/sasa.html), I have seen that there is only a very small difference or error between the analytic and numerical calculated per atom sasa "biggest difference in area between analytic and approximate calculations differing by 0.5 to 2% of full atom sphere area", so I think I will run with that.
The MSMS solvent excluded surface calculation in Chimera failed on many large atomic models (> 10,000 atoms) so we replaced it with a grid based solvent excluded surface algorithm which reliably computes surfaces for any size atomic model.[1]
That is interesting and probably explains why ChimeraX outputs easily 5 times more vertices, that is, can handle much higher vertices densities than MSMS can. The new algorithm for surface calculation is mentioned in the 2018 paper, however, it is not described in detail. Is there a separate piece of work dedicated to this new method of calculating molecular surfaces. I am wondering why MSMS is still often used and seen in other programs that use it as a utility library. Probably MSMS is very much established, robust (for not too large molecules) and well tested I guess and thus widley used. Is there a standalone program to use the surface calculation employed by ChimeraX? I am just curious, so far I just run ChimeraX from the command-line and let it calculate the surface for each pdb file using a simple script with "FOREACHFILE". This is just fine, but for more seamless integration in other scripts or programs a standalone software such as MSMS might be useful. Btw, as we are talking about it, when rendering surfaces at lower resolution by adding Gaussians centered at the atoms with appropriate contouring, shouldn't one be able to approximate the appearance of a SAS. Technically, lower resolution is not the same as a true SAS, but SAS will also result in smoothing of the SES. For calculation of course one uses "measure sasa" employing the exact analytic calculation, but for visual SAS representation, I am wondering how much of a legit approximation that would be? Any way, thank you again to both of you ( and the team ;) ) for all you great work. Cheers Dennis Quoting Tom Goddard <goddard@sonic.net>:
ChimeraX knows which atom every surface vertex is associated with. But if you just want the solvent accessible surface area per atom then use the "measure sasa" command as Elaine suggested. The per-atom area calculation does not use the displayed triangles. Instead it does an analytic calculation that is described here [1] The MSMS solvent excluded surface calculation in Chimera failed on many large atomic models (> 10,000 atoms) so we replaced it with a grid based solvent excluded surface algorithm which reliably computes surfaces for any size atomic model.[1] [1] Tom[1]
On Oct 19, 2023, at 9:55 AM, Elaine Meng via ChimeraX-users <chimerax-users@cgl.ucsf.edu> wrote:[1]
Hi Dennis, If you use the "measure sasa" command in ChimeraX, you can assign SASA values per atom and residue as attributes named "area":
<https://rbvi.ucsf.edu/chimerax/docs/user/commands/measure.html#sasa>
Attributes can be used for specification, coloring, etc., and written to a file with "save"
<https://rbvi.ucsf.edu/chimerax/docs/user/attributes.html> <https://rbvi.ucsf.edu/chimerax/docs/user/commands/save.html#attributes>
SESA is not available, however. ChimeraX does use a different method than Chimera to calculate the molecular surface, much more robust (less prone to failures).
<https://rbvi.ucsf.edu/chimerax/docs/user/commands/surface.html>
I hope this helps, Elaine ----- Elaine C. Meng, Ph.D. UCSF Chimera(X) team Department of Pharmaceutical Chemistry University of California, San Francisco[1]
On Oct 19, 2023, at 4:33 AM, Dennis Dannecker via ChimeraX-users <chimerax-users@cgl.ucsf.edu> wrote:
Hello Chimera/ChimeraX team
I recently asked already a question regarding the saving/exporting of vertices generated during the surface calculation in ChimeraX and the associated parameters with this surface generation.
On a related note, I wanted to ask whether it is possible to also extract the surface (SESA and/or SASA) on a per atom basis. MSMS calculates not only the vertices for each atom (usually several vertices per atom) but it also calculates the SESA and SASA on a per atom basis. Now, I was wondering whether something is possible in ChimeraX too. As I was parsing through the docs, I was wondering whether ChimeraX calculates the surface no longer using MSMS. The vertices density supported by ChimeraX is considerably higher than what I can get with my standalone MSMS program (at least factor 6). So, if ChimeraX does not longer employ MSMS, what else does it use?
With "measure convexity" I get the surface vertex positions, normals, convexity values, and triangles. Unfortunately, the association with the corresponding residue is lost (however, can be regained with a little bit of work I guess, simply using euclidean distance).
Question: Is there something similar for the SESA or SASA of the different atoms in the model/chain. In theory, this information should be available, at least the SESA. I am just wondering whether there is a way to write this out as a raw txt file in ChimeraX
The closest I found in the save command is the .obj or .vtk format. However, both are semi-satisfying in that that they both do not provide the data looked for and are in somewhat special formats, at least the vtk. The .obj file is much much bigger than what MSMS or "measure convexity" generates, and not really handy for then working with the data in other programs, mainly python.
As always, I very much appreciate any help and wish to thank the team for their contributions to the field.
Best regards
Dennis[1]
_______________________________________________ ChimeraX-users mailing list -- chimerax-users@cgl.ucsf.edu To unsubscribe send an email to chimerax-users-leave@cgl.ucsf.edu Archives: https://mail.cgl.ucsf.edu/mailman/archives/list/chimerax-users@cgl.ucsf.edu/ [1]
Links: ------ [1] https://www.cgl.ucsf.edu/chimera/data/sasa-nov2013/<span%20index=
Hi Dennis, I would only add that you could certainly make a visual comparison between the Gaussian and "standard" molecular surfaces by opening some structure twice, making each kind of surface on one copy, and then changing the display to transparent (one surface only, not both) and/or mesh. One could probably also devise some kind of quantitative comparison, but it would be more work, and probably you could sufficiently satisfy your curiousity about the magnitude of differences with your chosen set of parameters by just doing this visual comparison on one or two structures. Best, Elaine ----- Elaine C. Meng, Ph.D. UCSF Chimera(X) team Department of Pharmaceutical Chemistry University of California, San Francisco
On Oct 20, 2023, at 6:08 AM, Dennis Dannecker via ChimeraX-users <chimerax-users@cgl.ucsf.edu> wrote:
Hello Tom,
thank you very much for your reply.
Since you were saying, "ChimeraX knows which atom every surface vertex is associated with." , as a matter of fact, I would like to have this vertex-atom assignment. If there is an easy way to write this out as a file, then I would definitely go with that. As of now, I do the assignment with simply finding the closest atom for each vertex with a small script, it is reasonably fast thanks to vectorization and multiprocessing. But more elegant of cours ewould be to use what ChimeraX has already calculated.
I have realized that the calculated surface area per atom is not affected by the gridSpacing parameter used for surface calculation. Now that I know that ChimeraX uses an analytic calculation that independence makes of course sense.
Having read your work on the SASA from Nov 2013 (https://www.cgl.ucsf.edu/chimera/data/sasa-nov2013/sasa.html), I have seen that there is only a very small difference or error between the analytic and numerical calculated per atom sasa "biggest difference in area between analytic and approximate calculations differing by 0.5 to 2% of full atom sphere area", so I think I will run with that.
The MSMS solvent excluded surface calculation in Chimera failed on many large atomic models (> 10,000 atoms) so we replaced it with a grid based solvent excluded surface algorithm which reliably computes surfaces for any size atomic model.
That is interesting and probably explains why ChimeraX outputs easily 5 times more vertices, that is, can handle much higher vertices densities than MSMS can.
The new algorithm for surface calculation is mentioned in the 2018 paper, however, it is not described in detail. Is there a separate piece of work dedicated to this new method of calculating molecular surfaces. I am wondering why MSMS is still often used and seen in other programs that use it as a utility library. Probably MSMS is very much established, robust (for not too large molecules) and well tested I guess and thus widley used. Is there a standalone program to use the surface calculation employed by ChimeraX? I am just curious, so far I just run ChimeraX from the command-line and let it calculate the surface for each pdb file using a simple script with "forEachFile". This is just fine, but for more seamless integration in other scripts or programs a standalone software such as MSMS might be useful.
Btw, as we are talking about it, when rendering surfaces at lower resolution by adding Gaussians centered at the atoms with appropriate contouring, shouldn't one be able to approximate the appearance of a SAS. Technically, lower resolution is not the same as a true SAS, but SAS will also result in smoothing of the SES. For calculation of course one uses "measure sasa" employing the exact analytic calculation, but for visual SAS representation, I am wondering how much of a legit approximation that would be?
Any way, thank you again to both of you ( and the team ;) ) for all you great work.
Cheers
Dennis
Quoting Tom Goddard <goddard@sonic.net>:
ChimeraX knows which atom every surface vertex is associated with. But if you just want the solvent accessible surface area per atom then use the "measure sasa" command as Elaine suggested. The per-atom area calculation does not use the displayed triangles. Instead it does an analytic calculation that is described here
The MSMS solvent excluded surface calculation in Chimera failed on many large atomic models (> 10,000 atoms) so we replaced it with a grid based solvent excluded surface algorithm which reliably computes surfaces for any size atomic model.
Tom
On Oct 19, 2023, at 9:55 AM, Elaine Meng via ChimeraX-users <chimerax-users@cgl.ucsf.edu> wrote:
Hi Dennis, If you use the "measure sasa" command in ChimeraX, you can assign SASA values per atom and residue as attributes named "area":
<https://rbvi.ucsf.edu/chimerax/docs/user/commands/measure.html#sasa>
Attributes can be used for specification, coloring, etc., and written to a file with "save"
<https://rbvi.ucsf.edu/chimerax/docs/user/attributes.html> <https://rbvi.ucsf.edu/chimerax/docs/user/commands/save.html#attributes>
SESA is not available, however. ChimeraX does use a different method than Chimera to calculate the molecular surface, much more robust (less prone to failures).
<https://rbvi.ucsf.edu/chimerax/docs/user/commands/surface.html>
I hope this helps, Elaine ----- Elaine C. Meng, Ph.D. UCSF Chimera(X) team Department of Pharmaceutical Chemistry University of California, San Francisco
On Oct 19, 2023, at 4:33 AM, Dennis Dannecker via ChimeraX-users <chimerax-users@cgl.ucsf.edu> wrote:
Hello Chimera/ChimeraX team
I recently asked already a question regarding the saving/exporting of vertices generated during the surface calculation in ChimeraX and the associated parameters with this surface generation.
On a related note, I wanted to ask whether it is possible to also extract the surface (SESA and/or SASA) on a per atom basis. MSMS calculates not only the vertices for each atom (usually several vertices per atom) but it also calculates the SESA and SASA on a per atom basis. Now, I was wondering whether something is possible in ChimeraX too. As I was parsing through the docs, I was wondering whether ChimeraX calculates the surface no longer using MSMS. The vertices density supported by ChimeraX is considerably higher than what I can get with my standalone MSMS program (at least factor 6). So, if ChimeraX does not longer employ MSMS, what else does it use?
With "measure convexity" I get the surface vertex positions, normals, convexity values, and triangles. Unfortunately, the association with the corresponding residue is lost (however, can be regained with a little bit of work I guess, simply using euclidean distance).
Question: Is there something similar for the SESA or SASA of the different atoms in the model/chain. In theory, this information should be available, at least the SESA. I am just wondering whether there is a way to write this out as a raw txt file in ChimeraX
The closest I found in the save command is the .obj or .vtk format. However, both are semi-satisfying in that that they both do not provide the data looked for and are in somewhat special formats, at least the vtk. The .obj file is much much bigger than what MSMS or "measure convexity" generates, and not really handy for then working with the data in other programs, mainly python.
As always, I very much appreciate any help and wish to thank the team for their contributions to the field.
Best regards
Dennis
Hi Dennis, You can access all the surface information from Python. For example, I see in the ChimeraX programming manual that MolecularSurface has a vertex_to_atom_map() method that gives the atom index (starting at 0) for each vertex. Here's how I could write that to a file. First open an atomic model and make a surface open 1a0m surface Now in the ChimeraX Python shell, menu Tools / General / Shell, I could type from chimerax.atomic import MolecularSurface for s in session.models.list(type = MolecularSurface): with open(s.name + '.txt', 'w') as f: for a in s.vertex_to_atom_map(): f.write(f'{a}\n') which loops over all molecular surfaces (there are two since 1a0m has 2 chains), and writes files (1a0m_A SES surface.txt, 1a0m_B SES surface.txt) that contain as many lines as there are surface vertices (13441 for chain A surface) with the index of the atom for that vertex on that line.
1a0m_A SES surface.txt 94 94 ... 21 20 17
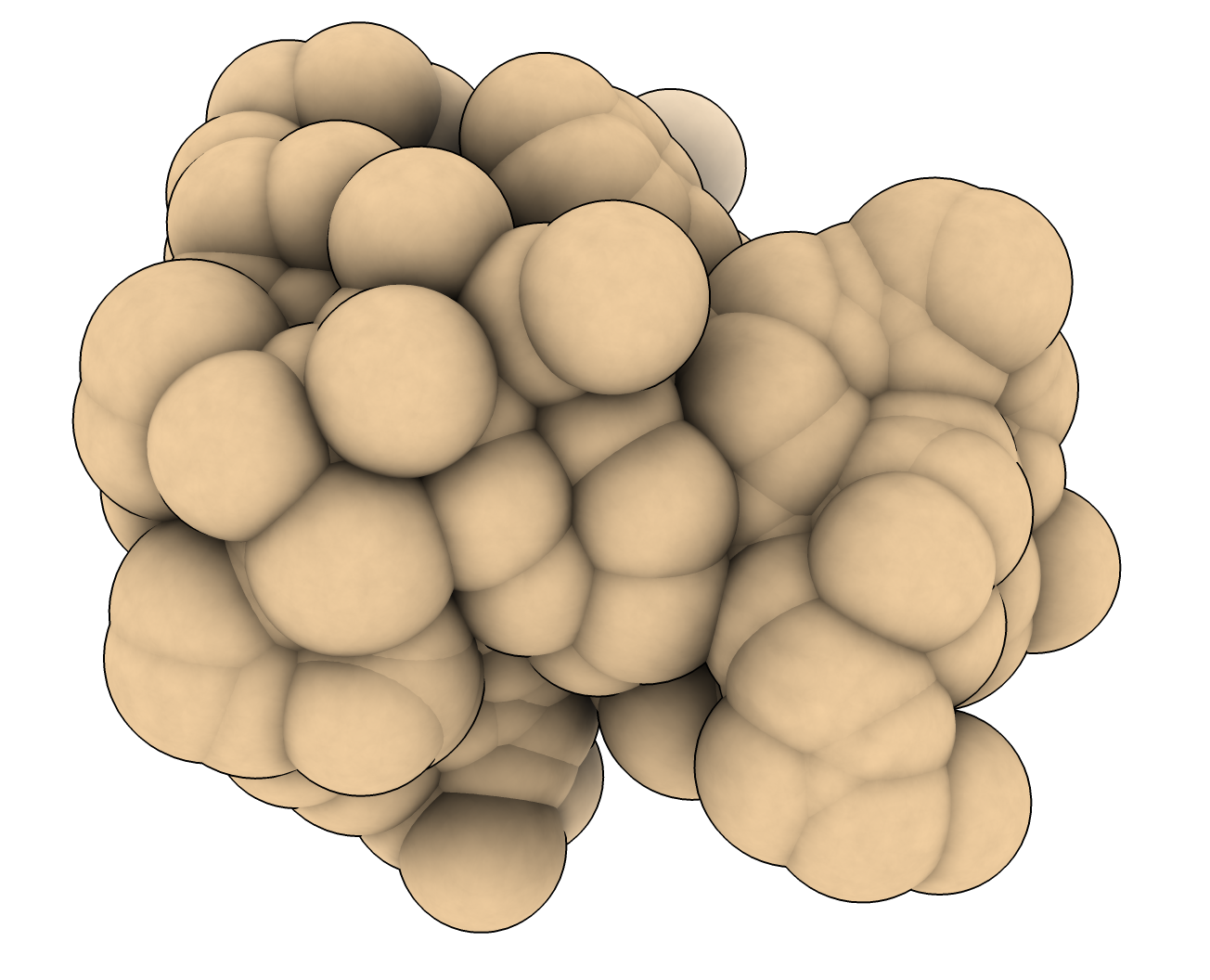
There is no article on the ChimeraX molecular surface algorithm, nor is there a standalone program to use it. It is a combination of Python and C++ code. Here is the Python https://github.com/RBVI/ChimeraX/blob/375a87d393e0f137a96cae9592b8112f1ad9ba... and you can see that it uses the Python numpy array package and imports some ChimeraX C++ routines like sphere_surface_distance(), contour_surface(), connected_pieces(), find_closest_points() optimized for speed. These routines are all part of the ChimeraX library that just was released for Mac on PyPi https://pypi.org/project/ChimeraX/ Using that you could write your own Python code that uses this library, or you could just use the ChimeraX application in "nogui" mode to run the calculations. If you want to see what a solvent accessible surface looks like, just show the atoms as spheres and increase all their radii by the probe radius (e.g. 1.4 Angstroms, command "size atomRadius +.1)). Does that look like a Gaussian surface? Only in the vaguest sense that it has the shape of the molecule. Here's an image of the SAS surface for PDB 1a0m. Tom 
On Oct 20, 2023, at 6:08 AM, Dennis Dannecker via ChimeraX-users <chimerax-users@cgl.ucsf.edu> wrote:
Hello Tom,
thank you very much for your reply.
Since you were saying, "ChimeraX knows which atom every surface vertex is associated with." , as a matter of fact, I would like to have this vertex-atom assignment. If there is an easy way to write this out as a file, then I would definitely go with that. As of now, I do the assignment with simply finding the closest atom for each vertex with a small script, it is reasonably fast thanks to vectorization and multiprocessing. But more elegant of cours ewould be to use what ChimeraX has already calculated.
I have realized that the calculated surface area per atom is not affected by the gridSpacing parameter used for surface calculation. Now that I know that ChimeraX uses an analytic calculation that independence makes of course sense.
Having read your work on the SASA from Nov 2013 (https://www.cgl.ucsf.edu/chimera/data/sasa-nov2013/sasa.html), I have seen that there is only a very small difference or error between the analytic and numerical calculated per atom sasa "biggest difference in area between analytic and approximate calculations differing by 0.5 to 2% of full atom sphere area", so I think I will run with that.
The MSMS solvent excluded surface calculation in Chimera failed on many large atomic models (> 10,000 atoms) so we replaced it with a grid based solvent excluded surface algorithm which reliably computes surfaces for any size atomic model. <https://www.cgl.ucsf.edu/chimera/data/sasa-nov2013/%3Cspan%20index=>
That is interesting and probably explains why ChimeraX outputs easily 5 times more vertices, that is, can handle much higher vertices densities than MSMS can.
The new algorithm for surface calculation is mentioned in the 2018 paper, however, it is not described in detail. Is there a separate piece of work dedicated to this new method of calculating molecular surfaces. I am wondering why MSMS is still often used and seen in other programs that use it as a utility library. Probably MSMS is very much established, robust (for not too large molecules) and well tested I guess and thus widley used. Is there a standalone program to use the surface calculation employed by ChimeraX? I am just curious, so far I just run ChimeraX from the command-line and let it calculate the surface for each pdb file using a simple script with "forEachFile". This is just fine, but for more seamless integration in other scripts or programs a standalone software such as MSMS might be useful.
Btw, as we are talking about it, when rendering surfaces at lower resolution by adding Gaussians centered at the atoms with appropriate contouring, shouldn't one be able to approximate the appearance of a SAS. Technically, lower resolution is not the same as a true SAS, but SAS will also result in smoothing of the SES. For calculation of course one uses "measure sasa" employing the exact analytic calculation, but for visual SAS representation, I am wondering how much of a legit approximation that would be?
Any way, thank you again to both of you ( and the team ;) ) for all you great work.
Cheers
Dennis
Quoting Tom Goddard <goddard@sonic.net <mailto:goddard@sonic.net>>:
ChimeraX knows which atom every surface vertex is associated with. But if you just want the solvent accessible surface area per atom then use the "measure sasa" command as Elaine suggested. The per-atom area calculation does not use the displayed triangles. Instead it does an analytic calculation that is described here
<https://www.cgl.ucsf.edu/chimera/data/sasa-nov2013/%3Cspan%20index=> The MSMS solvent excluded surface calculation in Chimera failed on many large atomic models (> 10,000 atoms) so we replaced it with a grid based solvent excluded surface algorithm which reliably computes surfaces for any size atomic model. <https://www.cgl.ucsf.edu/chimera/data/sasa-nov2013/%3Cspan%20index=> <https://www.cgl.ucsf.edu/chimera/data/sasa-nov2013/%3Cspan%20index=> Tom <https://www.cgl.ucsf.edu/chimera/data/sasa-nov2013/%3Cspan%20index=>
On Oct 19, 2023, at 9:55 AM, Elaine Meng via ChimeraX-users <chimerax-users@cgl.ucsf.edu> wrote: <https://www.cgl.ucsf.edu/chimera/data/sasa-nov2013/%3Cspan%20index=> Hi Dennis, If you use the "measure sasa" command in ChimeraX, you can assign SASA values per atom and residue as attributes named "area":
<https://rbvi.ucsf.edu/chimerax/docs/user/commands/measure.html#sasa>
Attributes can be used for specification, coloring, etc., and written to a file with "save"
<https://rbvi.ucsf.edu/chimerax/docs/user/attributes.html> <https://rbvi.ucsf.edu/chimerax/docs/user/commands/save.html#attributes>
SESA is not available, however. ChimeraX does use a different method than Chimera to calculate the molecular surface, much more robust (less prone to failures).
<https://rbvi.ucsf.edu/chimerax/docs/user/commands/surface.html>
I hope this helps, Elaine ----- Elaine C. Meng, Ph.D. UCSF Chimera(X) team Department of Pharmaceutical Chemistry University of California, San Francisco <https://www.cgl.ucsf.edu/chimera/data/sasa-nov2013/%3Cspan%20index=>
On Oct 19, 2023, at 4:33 AM, Dennis Dannecker via ChimeraX-users <chimerax-users@cgl.ucsf.edu> wrote:
Hello Chimera/ChimeraX team
I recently asked already a question regarding the saving/exporting of vertices generated during the surface calculation in ChimeraX and the associated parameters with this surface generation.
On a related note, I wanted to ask whether it is possible to also extract the surface (SESA and/or SASA) on a per atom basis. MSMS calculates not only the vertices for each atom (usually several vertices per atom) but it also calculates the SESA and SASA on a per atom basis. Now, I was wondering whether something is possible in ChimeraX too. As I was parsing through the docs, I was wondering whether ChimeraX calculates the surface no longer using MSMS. The vertices density supported by ChimeraX is considerably higher than what I can get with my standalone MSMS program (at least factor 6). So, if ChimeraX does not longer employ MSMS, what else does it use?
With "measure convexity" I get the surface vertex positions, normals, convexity values, and triangles. Unfortunately, the association with the corresponding residue is lost (however, can be regained with a little bit of work I guess, simply using euclidean distance).
Question: Is there something similar for the SESA or SASA of the different atoms in the model/chain. In theory, this information should be available, at least the SESA. I am just wondering whether there is a way to write this out as a raw txt file in ChimeraX
The closest I found in the save command is the .obj or .vtk format. However, both are semi-satisfying in that that they both do not provide the data looked for and are in somewhat special formats, at least the vtk. The .obj file is much much bigger than what MSMS or "measure convexity" generates, and not really handy for then working with the data in other programs, mainly python.
As always, I very much appreciate any help and wish to thank the team for their contributions to the field.
Best regards
Dennis <https://www.cgl.ucsf.edu/chimera/data/sasa-nov2013/%3Cspan%20index=>
_______________________________________________ ChimeraX-users mailing list -- chimerax-users@cgl.ucsf.edu To unsubscribe send an email to chimerax-users-leave@cgl.ucsf.edu Archives: https://mail.cgl.ucsf.edu/mailman/archives/list/chimerax-users@cgl.ucsf.edu/ <https://www.cgl.ucsf.edu/chimera/data/sasa-nov2013/%3Cspan%20index=>
_______________________________________________ ChimeraX-users mailing list -- chimerax-users@cgl.ucsf.edu <mailto:chimerax-users@cgl.ucsf.edu> To unsubscribe send an email to chimerax-users-leave@cgl.ucsf.edu <mailto:chimerax-users-leave@cgl.ucsf.edu> Archives: https://mail.cgl.ucsf.edu/mailman/archives/list/chimerax-users@cgl.ucsf.edu/
{kind=link}
participants (3)
-
 Dennis Dannecker
Dennis Dannecker -
 Elaine Meng
Elaine Meng -
 Tom Goddard
Tom Goddard