How to get back to original view after running resfit

Hi, I am using resfit command. Is there a command or way to go back to the original view? Best, Rayees

Hi Rayees, Depends on exactly what you mean by original view, but you can delete 2D labels, show all atoms, and turn off map zoning via GUI actions or commands, e.g. commands: 2dlab delete show atoms volume unzone If you wanted how the structure looked when you first opened it (e.g. maybe it was shown as ribbons rather than atoms), then one way is command: preset "initial styles" "original look" If you wanted original positions/orientations, one way is command: view orient; view initial There are usually several different ways to do the same thing (menu or gui actions, or even different sets of commands with same net result). I'm giving commands because it is a lot less typing than describing the other ways. All of these commands have detailed help in the User Guide (menu: Help... User Guide). I hope this helps, Elaine ----- Elaine C. Meng, Ph.D. UCSF Chimera(X) team Department of Pharmaceutical Chemistry University of California, San Francisco
On Aug 17, 2022, at 5:05 PM, Rayees Mattoo via ChimeraX-users <chimerax-users@cgl.ucsf.edu> wrote:
Hi, I am using resfit command. Is there a command or way to go back to the original view?
Best, Rayees
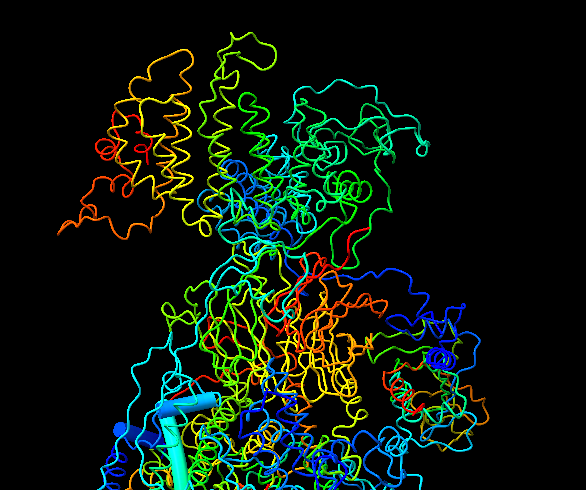
Hi Elaine, Thanks. That is exactly what I wanted. One more question. I built a model in coot and then I opened in chimeraX. The model in cartoon representation shows most of the helices and sheets as loops (snapshot attached). I was wondering if there is something wrong with my model or I need to do something in chimeraX to get the cartoon display right. Best Rayees ________________________________ From: Elaine Meng <meng@cgl.ucsf.edu> Sent: Wednesday, August 17, 2022 5:18 PM To: Rayees Mattoo <rmattoo@stanford.edu> Cc: ChimeraX Users Help <chimerax-users@cgl.ucsf.edu> Subject: Re: [chimerax-users] How to get back to original view after running resfit Hi Rayees, Depends on exactly what you mean by original view, but you can delete 2D labels, show all atoms, and turn off map zoning via GUI actions or commands, e.g. commands: 2dlab delete show atoms volume unzone If you wanted how the structure looked when you first opened it (e.g. maybe it was shown as ribbons rather than atoms), then one way is command: preset "initial styles" "original look" If you wanted original positions/orientations, one way is command: view orient; view initial There are usually several different ways to do the same thing (menu or gui actions, or even different sets of commands with same net result). I'm giving commands because it is a lot less typing than describing the other ways. All of these commands have detailed help in the User Guide (menu: Help... User Guide). I hope this helps, Elaine ----- Elaine C. Meng, Ph.D. UCSF Chimera(X) team Department of Pharmaceutical Chemistry University of California, San Francisco
On Aug 17, 2022, at 5:05 PM, Rayees Mattoo via ChimeraX-users <chimerax-users@cgl.ucsf.edu> wrote:
Hi, I am using resfit command. Is there a command or way to go back to the original view?
Best, Rayees
{kind=link}
Hi Rayees, I don't know what kind of file you saved from Coot and opened in ChimeraX. I'm guessing PDB. The strand and helix assignments are taken from the SHEET and HELIX records in the input PDB file, or if the PDB file doesn't have those, calculated automatically with "dssp" (take a look at the Log when you open the structure file, it would say if this was done). You could try re-running the "dssp" command with different parameter values, <https://rbvi.ucsf.edu/chimerax/docs/user/commands/dssp.html> or manually assign secondary structure by using the "setattr" command to assign values of the residue attribute named "ss_type" as 1 (helix) or 2 (strand) instead of 0 (coil): <https://rbvi.ucsf.edu/chimerax/docs/user/commands/setattr.html> <https://rbvi.ucsf.edu/chimerax/docs/user/attributes.html#residue> For example, to assign residues 10-20 of chain A as helix, command: setattr /A:10-20 r ss_type 1 However, there is another complication. It will still draw a thin (coil-like) connector between two residues if it thinks they are in DIFFERENT helices or strands from one another. So you may also need to assign "ss_id" values (in addition to "ss_type") to make sure that contigous residues of the same secondary structure type are also considered to be in the SAME secondary structure segment as one another. I hope this helps, Elaine ----- Elaine C. Meng, Ph.D. UCSF Chimera(X) team Department of Pharmaceutical Chemistry University of California, San Francisco
On Aug 17, 2022, at 6:23 PM, Rayees Mattoo via ChimeraX-users <chimerax-users@cgl.ucsf.edu> wrote:
Hi Elaine, Thanks. That is exactly what I wanted. One more question. I built a model in coot and then I opened in chimeraX. The model in cartoon representation shows most of the helices and sheets as loops (snapshot attached). I was wondering if there is something wrong with my model or I need to do something in chimeraX to get the cartoon display right.
Best Rayees
Great! Thanks. "dssp" solved the problem. Best Rayees ________________________________ From: Elaine Meng <meng@cgl.ucsf.edu> Sent: Wednesday, August 17, 2022 7:11 PM To: Rayees Mattoo <rmattoo@stanford.edu> Cc: ChimeraX Users Help <chimerax-users@cgl.ucsf.edu> Subject: assigning protein secondary structure (helix and strand) Hi Rayees, I don't know what kind of file you saved from Coot and opened in ChimeraX. I'm guessing PDB. The strand and helix assignments are taken from the SHEET and HELIX records in the input PDB file, or if the PDB file doesn't have those, calculated automatically with "dssp" (take a look at the Log when you open the structure file, it would say if this was done). You could try re-running the "dssp" command with different parameter values, <https://rbvi.ucsf.edu/chimerax/docs/user/commands/dssp.html> or manually assign secondary structure by using the "setattr" command to assign values of the residue attribute named "ss_type" as 1 (helix) or 2 (strand) instead of 0 (coil): <https://rbvi.ucsf.edu/chimerax/docs/user/commands/setattr.html> <https://rbvi.ucsf.edu/chimerax/docs/user/attributes.html#residue> For example, to assign residues 10-20 of chain A as helix, command: setattr /A:10-20 r ss_type 1 However, there is another complication. It will still draw a thin (coil-like) connector between two residues if it thinks they are in DIFFERENT helices or strands from one another. So you may also need to assign "ss_id" values (in addition to "ss_type") to make sure that contigous residues of the same secondary structure type are also considered to be in the SAME secondary structure segment as one another. I hope this helps, Elaine ----- Elaine C. Meng, Ph.D. UCSF Chimera(X) team Department of Pharmaceutical Chemistry University of California, San Francisco
On Aug 17, 2022, at 6:23 PM, Rayees Mattoo via ChimeraX-users <chimerax-users@cgl.ucsf.edu> wrote:
Hi Elaine, Thanks. That is exactly what I wanted. One more question. I built a model in coot and then I opened in chimeraX. The model in cartoon representation shows most of the helices and sheets as loops (snapshot attached). I was wondering if there is something wrong with my model or I need to do something in chimeraX to get the cartoon display right.
Best Rayees
participants (2)
-
 Elaine Meng
Elaine Meng -
 Rayees Mattoo
Rayees Mattoo